

The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells
- PMID: 33232666
- PMCID: PMC7858232
- DOI: 10.1016/j.cmet.2020.10.026
The Power of Plasticity-Metabolic Regulation of Hepatic Stellate Cells
Abstract
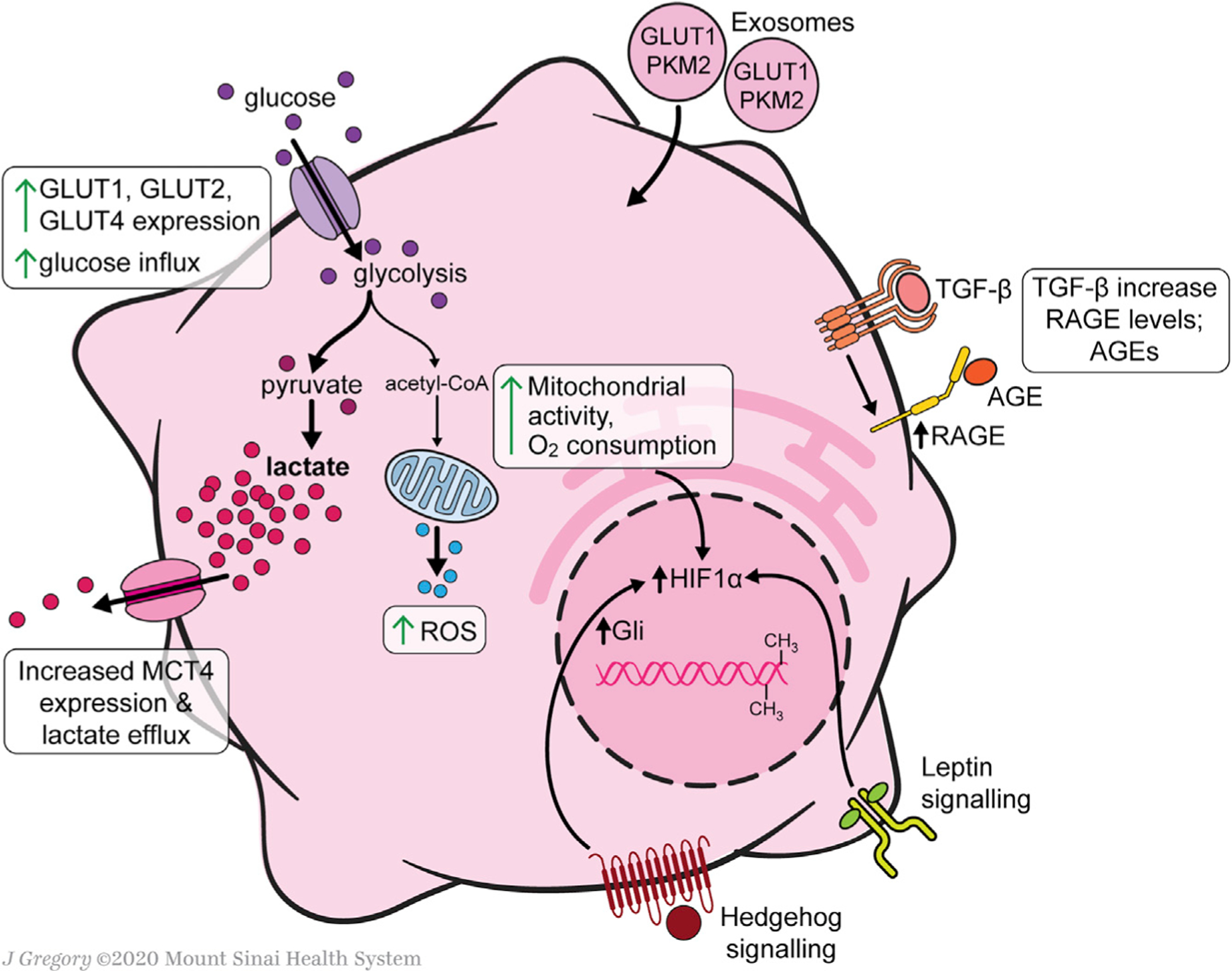
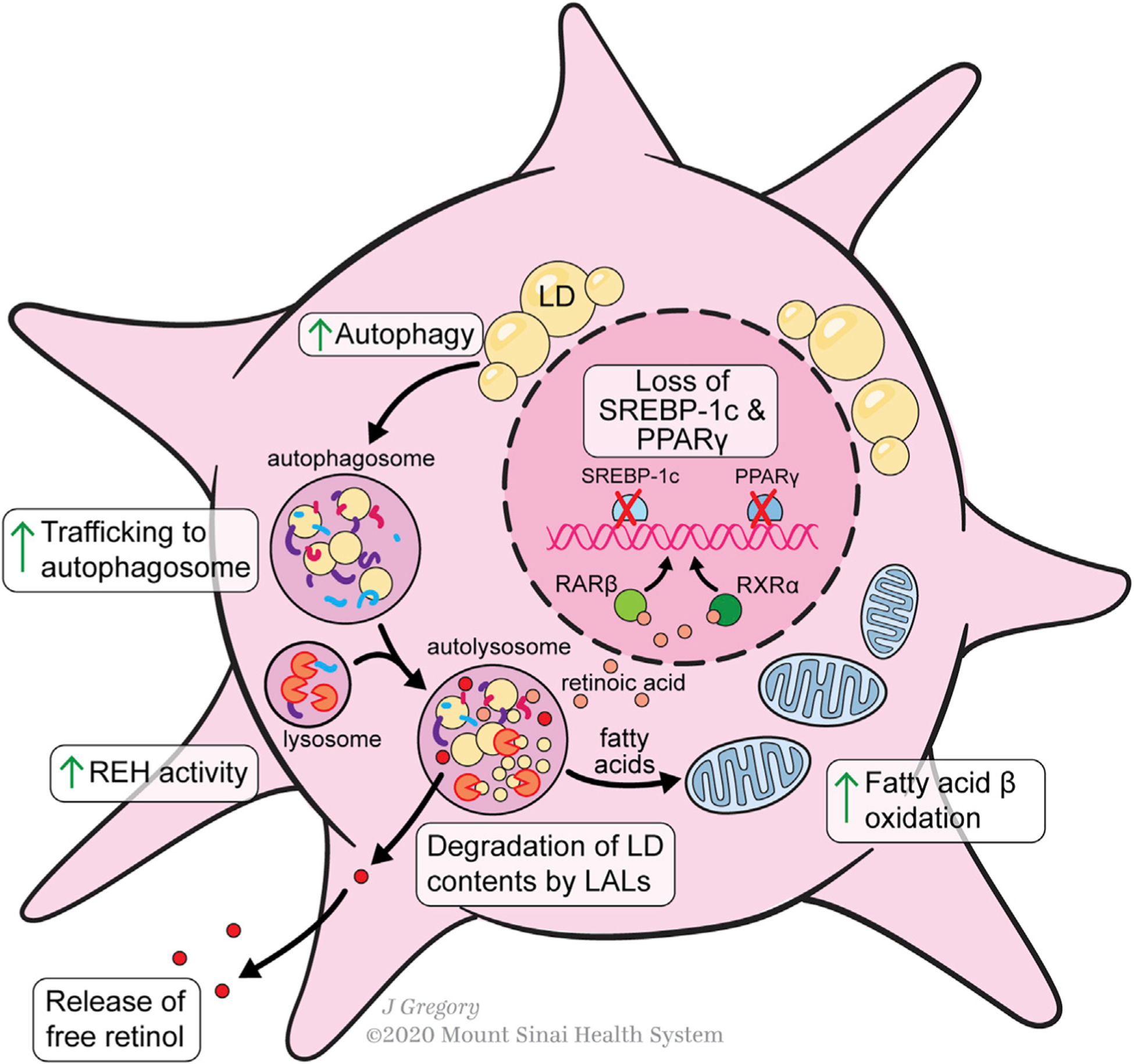
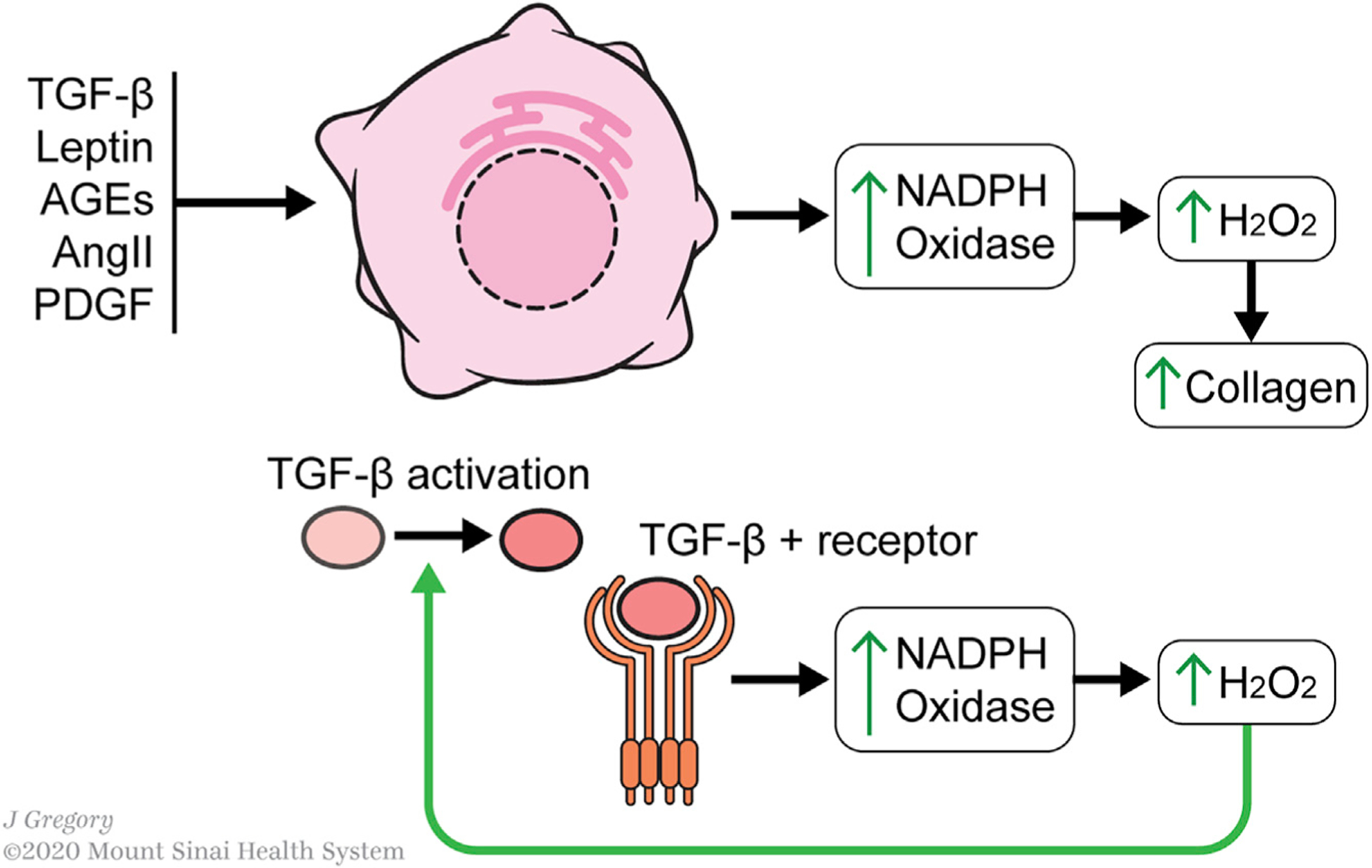
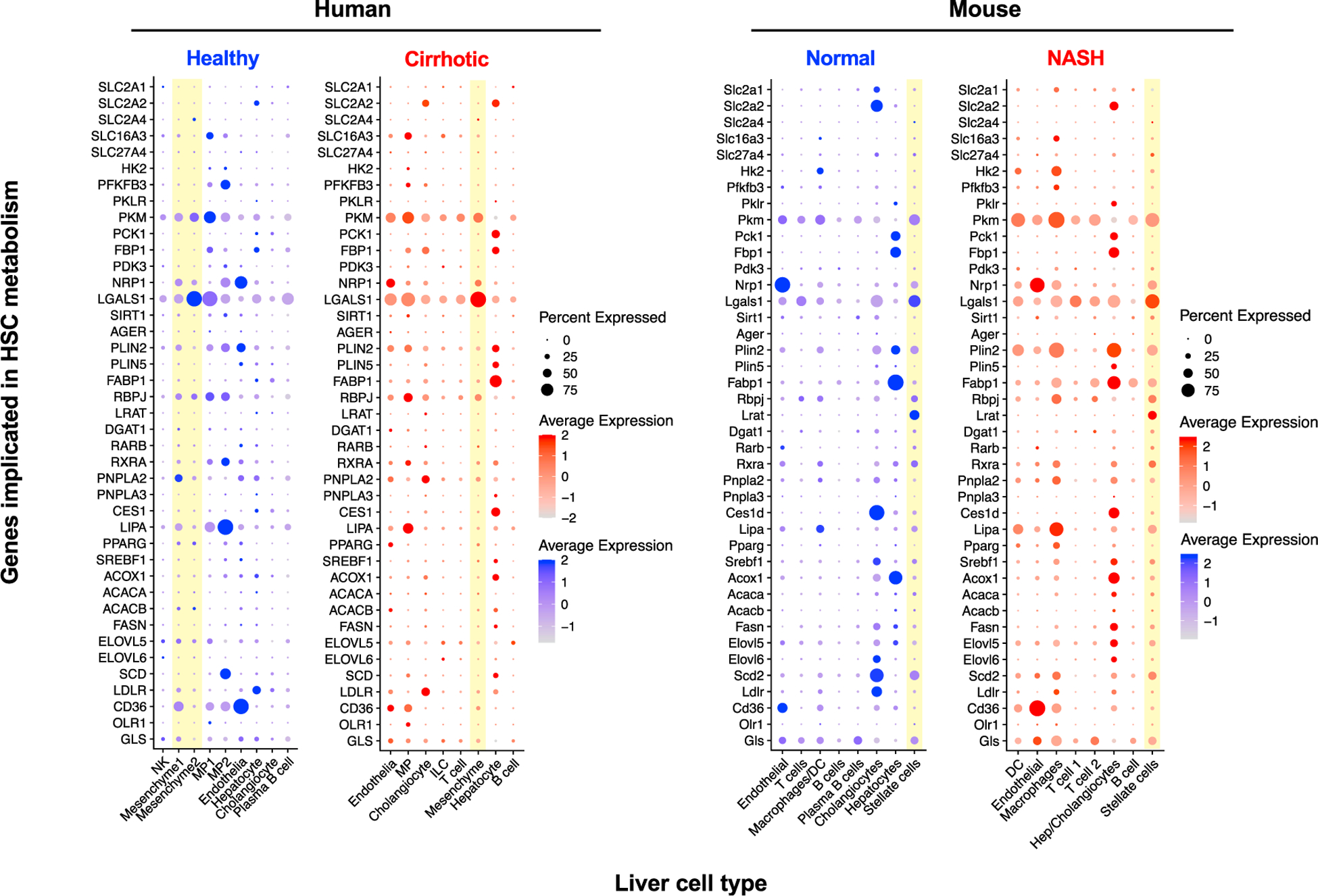
Hepatic stellate cells (HSCs) are resident non-parenchymal liver pericytes whose plasticity enables them to regulate a remarkable range of physiologic and pathologic responses. To support their functions in health and disease, HSCs engage pathways regulating carbohydrate, mitochondrial, lipid, and retinoid homeostasis. In chronic liver injury, HSCs drive hepatic fibrosis and are implicated in inflammation and cancer. To do so, the cells activate, or transdifferentiate, from a quiescent state into proliferative, motile myofibroblasts that secrete extracellular matrix, which demands rapid adaptation to meet a heightened energy need. Adaptations include reprogramming of central carbon metabolism, enhanced mitochondrial number and activity, endoplasmic reticulum stress, and liberation of free fatty acids through autophagy-dependent hydrolysis of retinyl esters that are stored in cytoplasmic droplets. As an archetype for pericytes in other tissues, recognition of the HSC's metabolic drivers and vulnerabilities offer the potential to target these pathways therapeutically to enhance parenchymal growth and modulate repair.
Copyright © 2020 Elsevier Inc. All rights reserved.
Figures

References
-
- Adachi T, Togashi H, Suzuki A, Kasai S, Ito J, Sugahara K, and Kawata S (2005). NAD(P)H oxidase plays a crucial role in PDGF-induced proliferation of hepatic stellate cells. Hepatology 41, 1272–1281. - PubMed
-
- Ajat M, Molenaar M, Brouwers JFHM, Vaandrager AB, Houweling M, and Helms JB (2017). Hepatic stellate cells retain the capacity to synthesize retinyl esters and to store neutral lipids in small lipid droplets in the absence of LRAT. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 176–187. - PubMed
-
- Amann T, and Hellerbrand C (2009). GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin. Ther. Targets 13, 1411–1427. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
