

Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells
- PMID: 33233489
- PMCID: PMC7699590
- DOI: 10.3390/ijms21228764
Reactive Oxygen Species: Modulators of Phenotypic Switch of Vascular Smooth Muscle Cells
Abstract
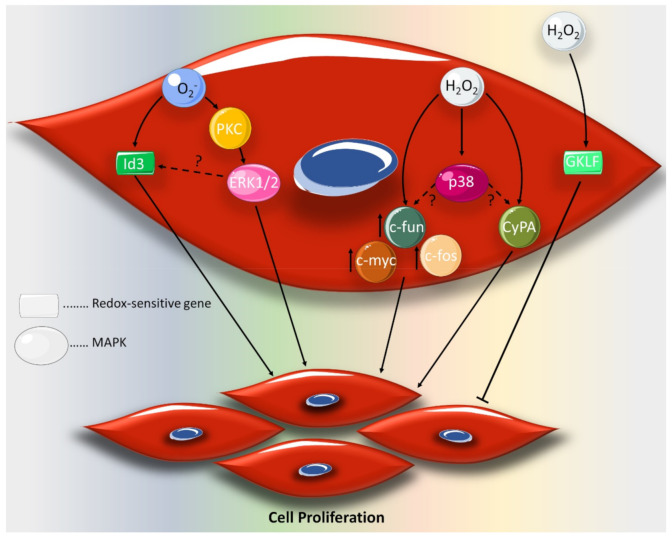
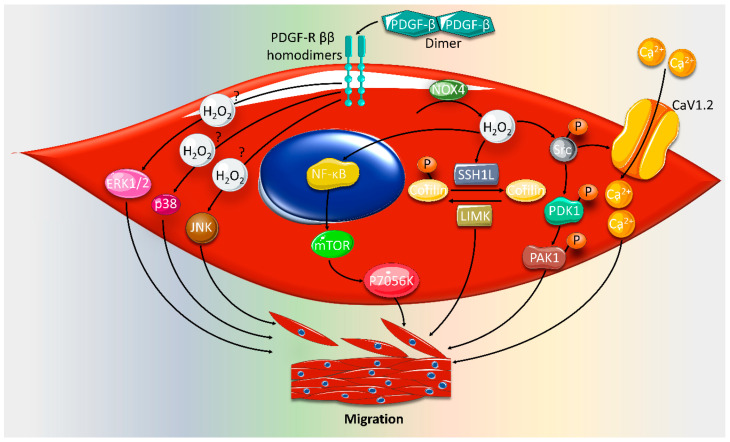
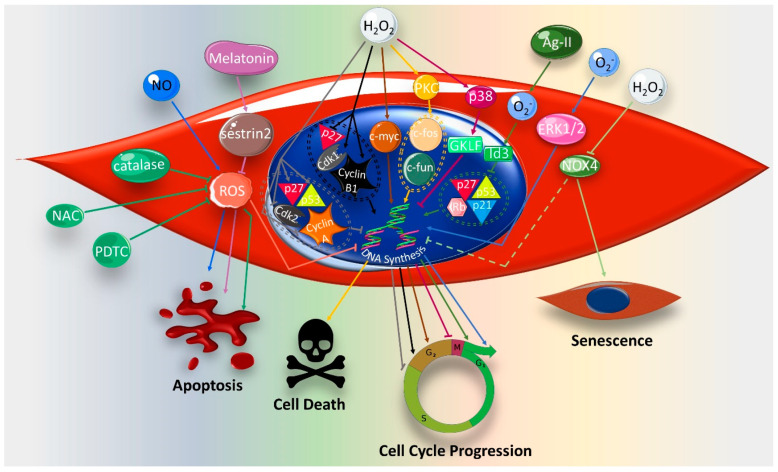
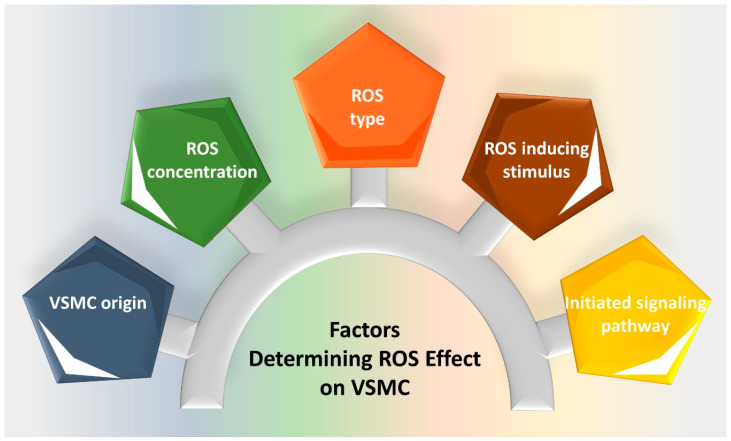
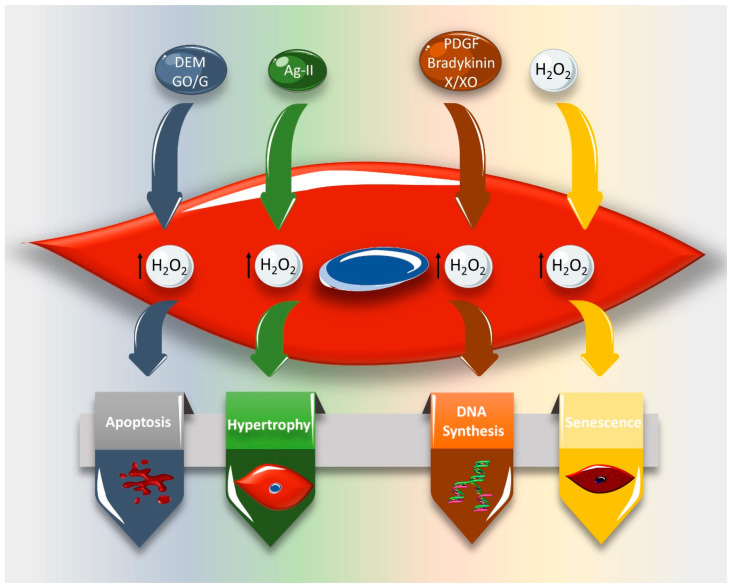
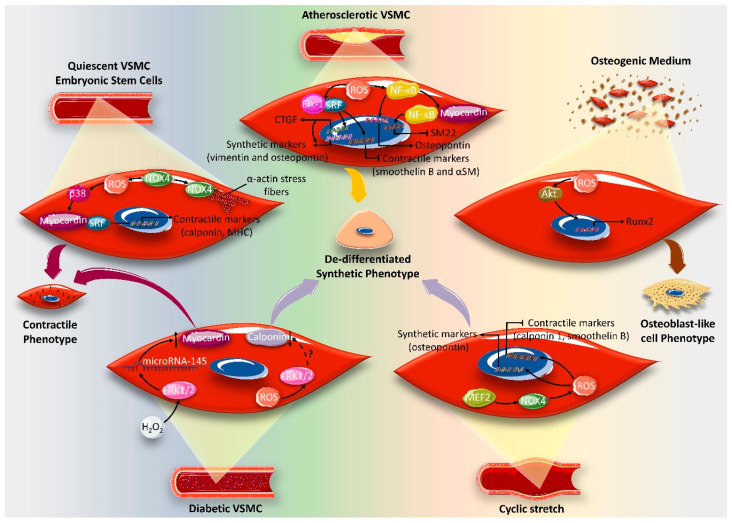
Reactive oxygen species (ROS) are natural byproducts of oxygen metabolism in the cell. At physiological levels, they play a vital role in cell signaling. However, high ROS levels cause oxidative stress, which is implicated in cardiovascular diseases (CVD) such as atherosclerosis, hypertension, and restenosis after angioplasty. Despite the great amount of research conducted to identify the role of ROS in CVD, the image is still far from being complete. A common event in CVD pathophysiology is the switch of vascular smooth muscle cells (VSMCs) from a contractile to a synthetic phenotype. Interestingly, oxidative stress is a major contributor to this phenotypic switch. In this review, we focus on the effect of ROS on the hallmarks of VSMC phenotypic switch, particularly proliferation and migration. In addition, we speculate on the underlying molecular mechanisms of these cellular events. Along these lines, the impact of ROS on the expression of contractile markers of VSMCs is discussed in depth. We conclude by commenting on the efficiency of antioxidants as CVD therapies.
Keywords: cardiovascular disease; phenotypic switch; reactive oxygen species; vascular smooth muscle cell.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Liu Y., Zhao H., Li H., Kalyanaraman B., Nicolosi A.C., Gutterman D.D. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ. Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
