

Understanding and exploiting interactions between cellular proteostasis pathways and infectious prion proteins for therapeutic benefit
- PMID: 33234071
- PMCID: PMC7729027
- DOI: 10.1098/rsob.200282
Understanding and exploiting interactions between cellular proteostasis pathways and infectious prion proteins for therapeutic benefit
Abstract
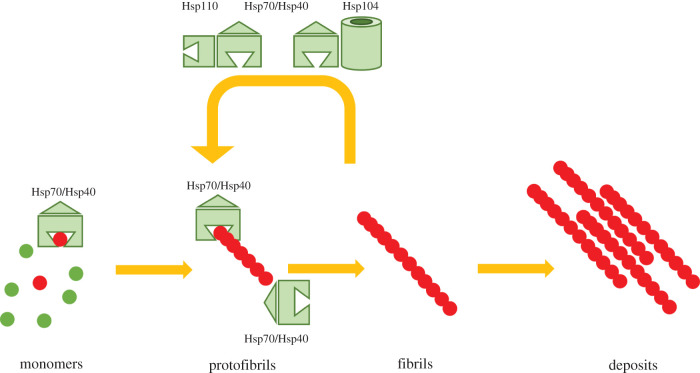
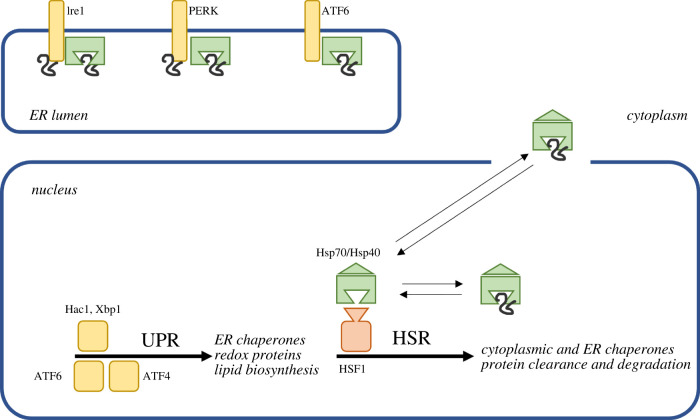
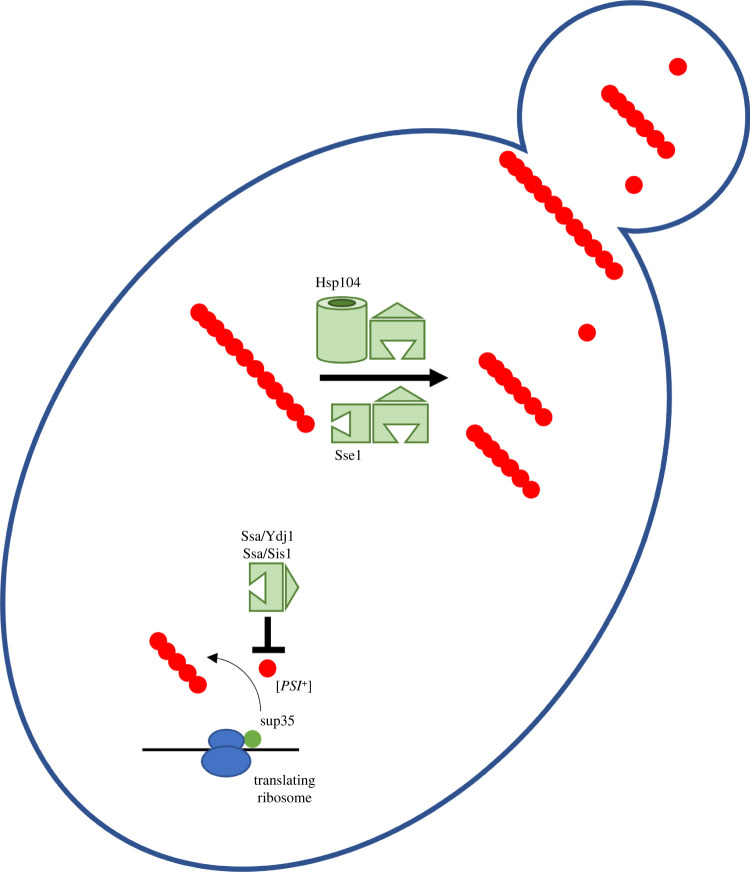
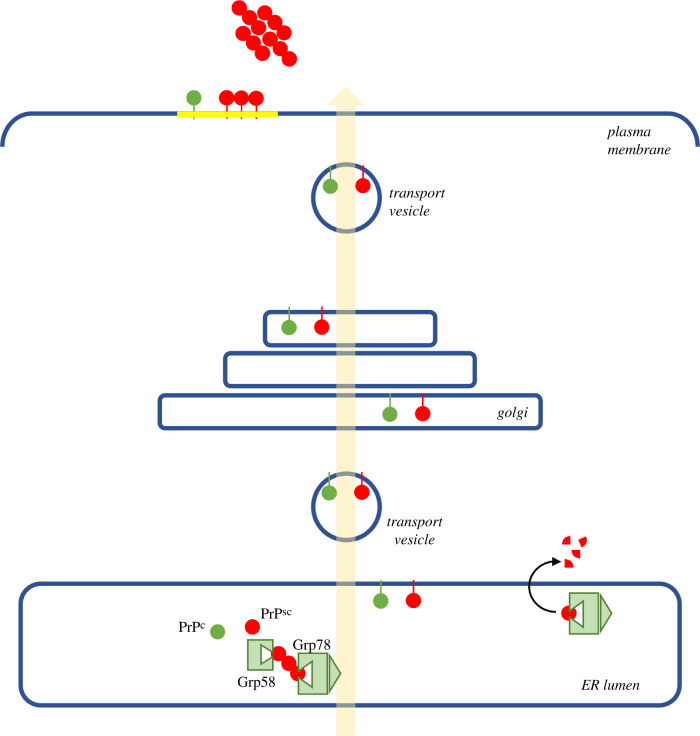
Several neurodegenerative diseases of humans and animals are caused by the misfolded prion protein (PrPSc), a self-propagating protein infectious agent that aggregates into oligomeric, fibrillar structures and leads to cell death by incompletely understood mechanisms. Work in multiple biological model systems, from simple baker's yeast to transgenic mouse lines, as well as in vitro studies, has illuminated molecular and cellular modifiers of prion disease. In this review, we focus on intersections between PrP and the proteostasis network, including unfolded protein stress response pathways and roles played by the powerful regulators of protein folding known as protein chaperones. We close with analysis of promising therapeutic avenues for treatment enabled by these studies.
Keywords: human; prions; protein chaperones; protein misfolding; stress; yeast.
Conflict of interest statement
We declare we have no competing interests.
Figures
Similar articles
-
Methionine oxidation within the prion protein.Prion. 2020 Dec;14(1):193-205. doi: 10.1080/19336896.2020.1796898. Prion. 2020. PMID: 32744136 Free PMC article. Review.
-
Anti-prion Protein Antibody 6D11 Restores Cellular Proteostasis of Prion Protein Through Disrupting Recycling Propagation of PrPSc and Targeting PrPSc for Lysosomal Degradation.Mol Neurobiol. 2019 Mar;56(3):2073-2091. doi: 10.1007/s12035-018-1208-4. Epub 2018 Jul 9. Mol Neurobiol. 2019. PMID: 29987703 Free PMC article.
-
In Vitro Approach To Identify Key Amino Acids in Low Susceptibility of Rabbit Prion Protein to Misfolding.J Virol. 2017 Nov 30;91(24):e01543-17. doi: 10.1128/JVI.01543-17. Print 2017 Dec 15. J Virol. 2017. PMID: 28978705 Free PMC article.
-
Molecular Mechanism of the Misfolding and Oligomerization of the Prion Protein: Current Understanding and Its Implications.Biochemistry. 2015 Jul 28;54(29):4431-42. doi: 10.1021/acs.biochem.5b00605. Epub 2015 Jul 17. Biochemistry. 2015. PMID: 26171558 Review.
-
Transition of the prion protein from a structured cellular form (PrPC ) to the infectious scrapie agent (PrPSc ).Protein Sci. 2019 Dec;28(12):2055-2063. doi: 10.1002/pro.3735. Epub 2019 Oct 25. Protein Sci. 2019. PMID: 31583788 Free PMC article. Review.
Cited by
-
Suppression of aggregate and amyloid formation by a novel intrinsically disordered region in metazoan Hsp110 chaperones.J Biol Chem. 2021 Jan-Jun;296:100567. doi: 10.1016/j.jbc.2021.100567. Epub 2021 Mar 19. J Biol Chem. 2021. PMID: 33753171 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
