

Mitochondrial quality control in kidney injury and repair
- PMID: 33235391
- PMCID: PMC8958893
- DOI: 10.1038/s41581-020-00369-0
Mitochondrial quality control in kidney injury and repair
Abstract
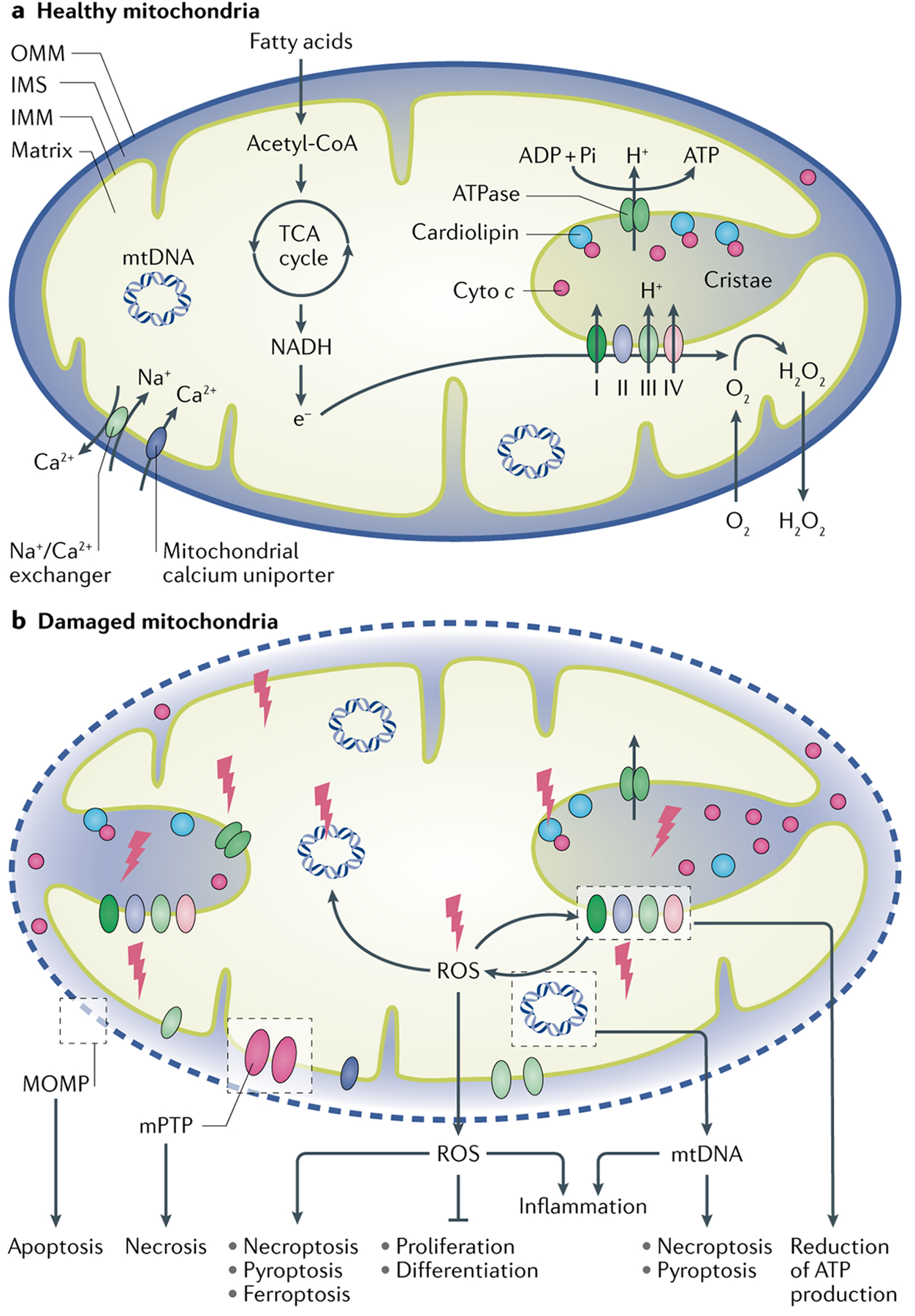
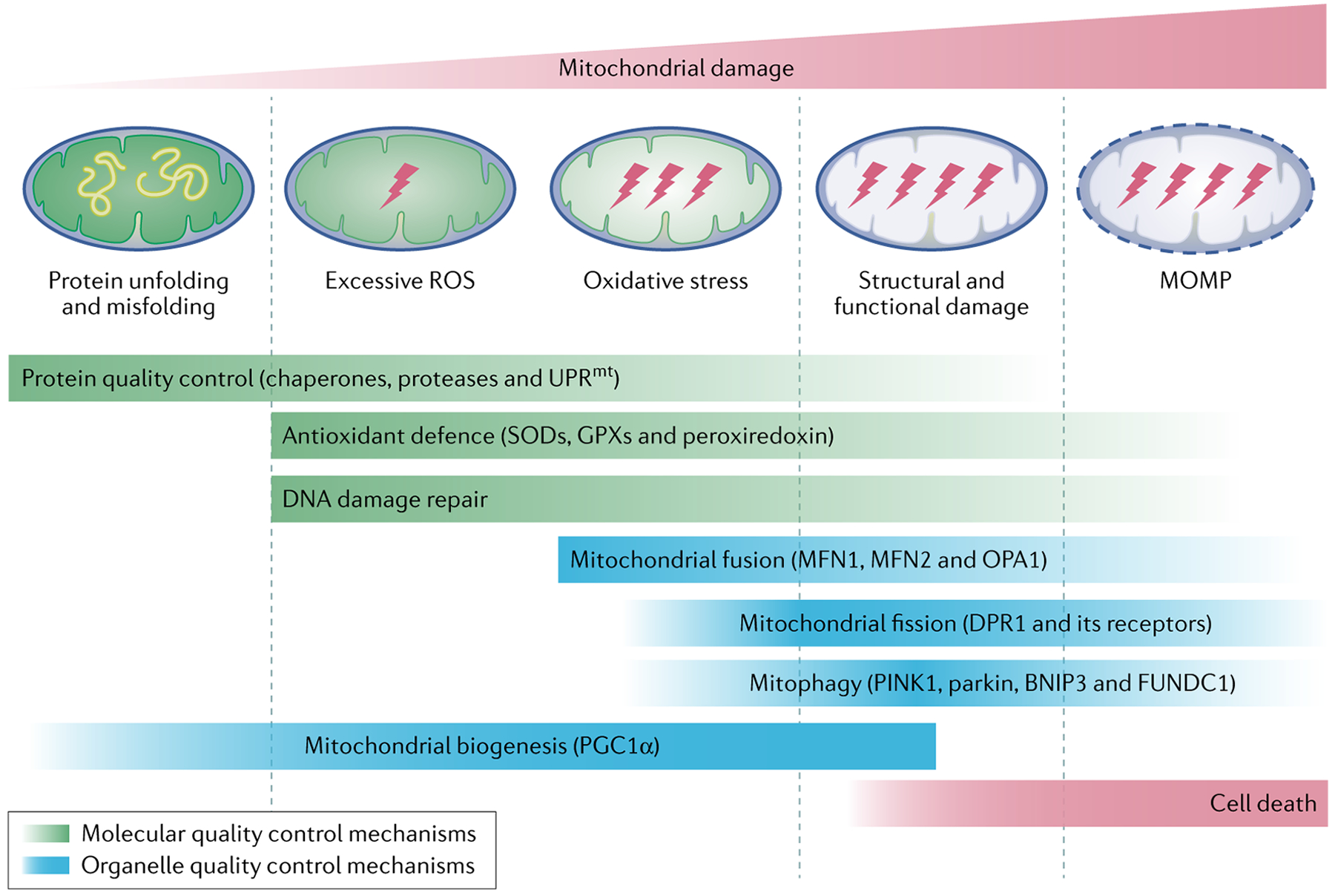
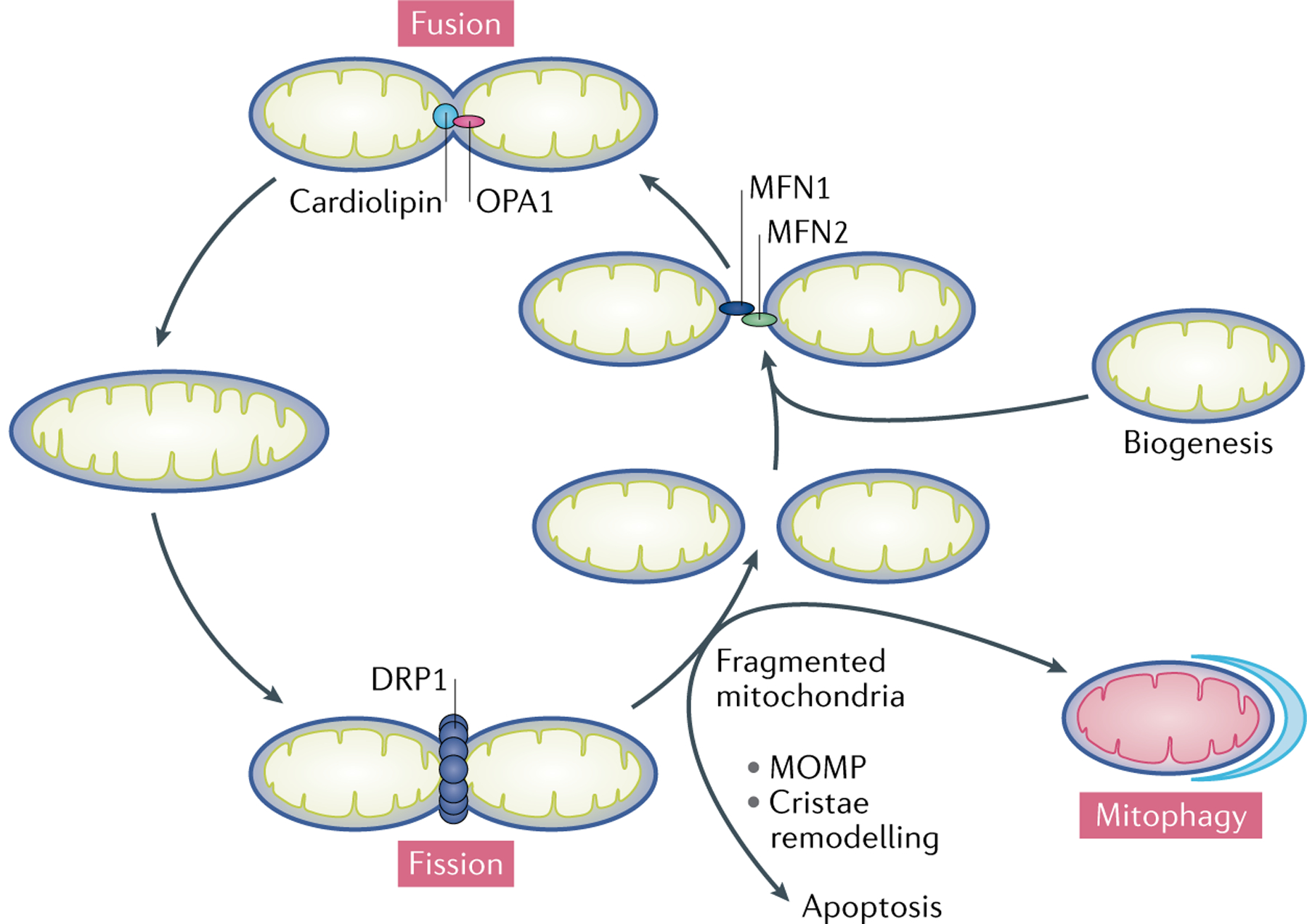
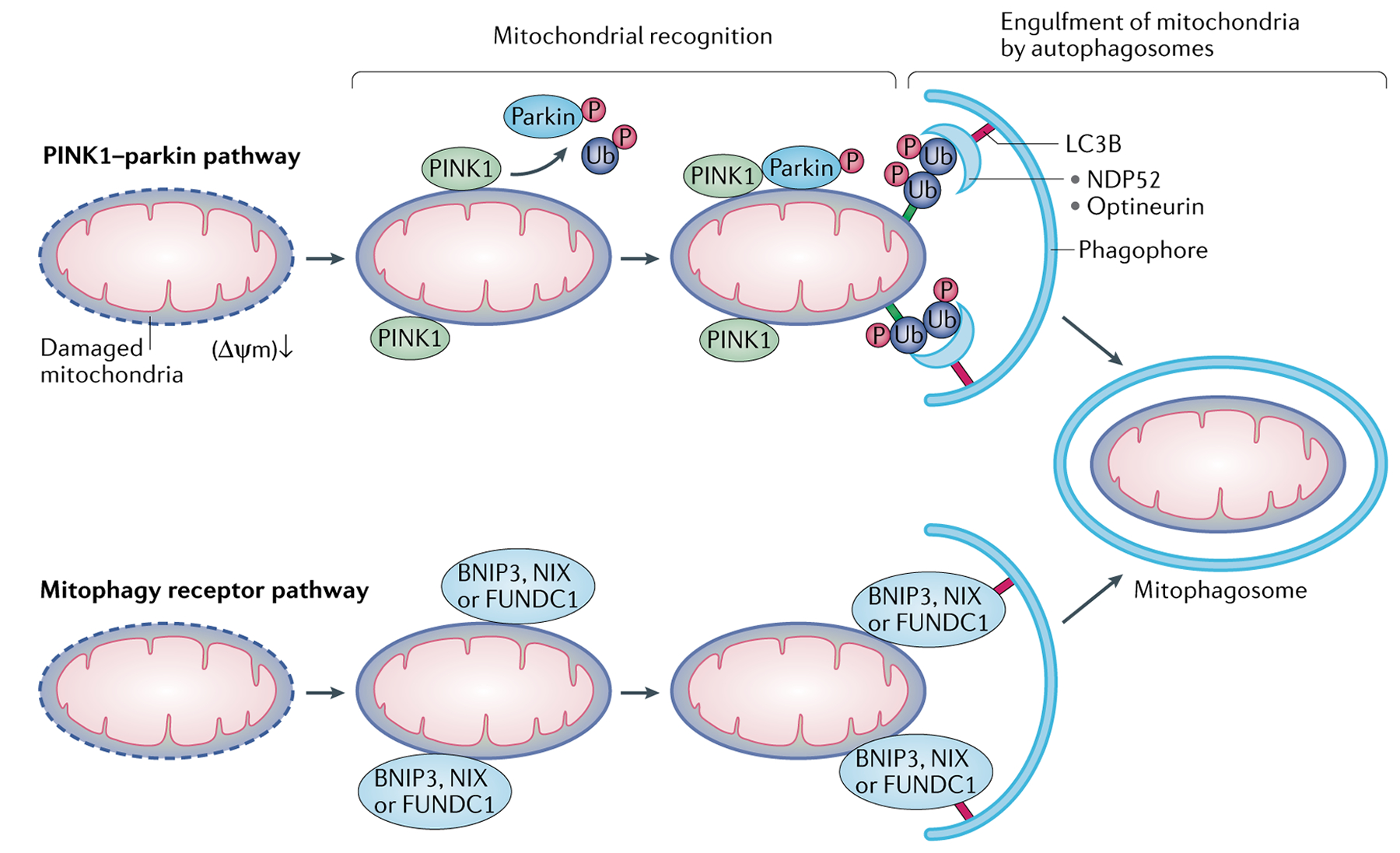
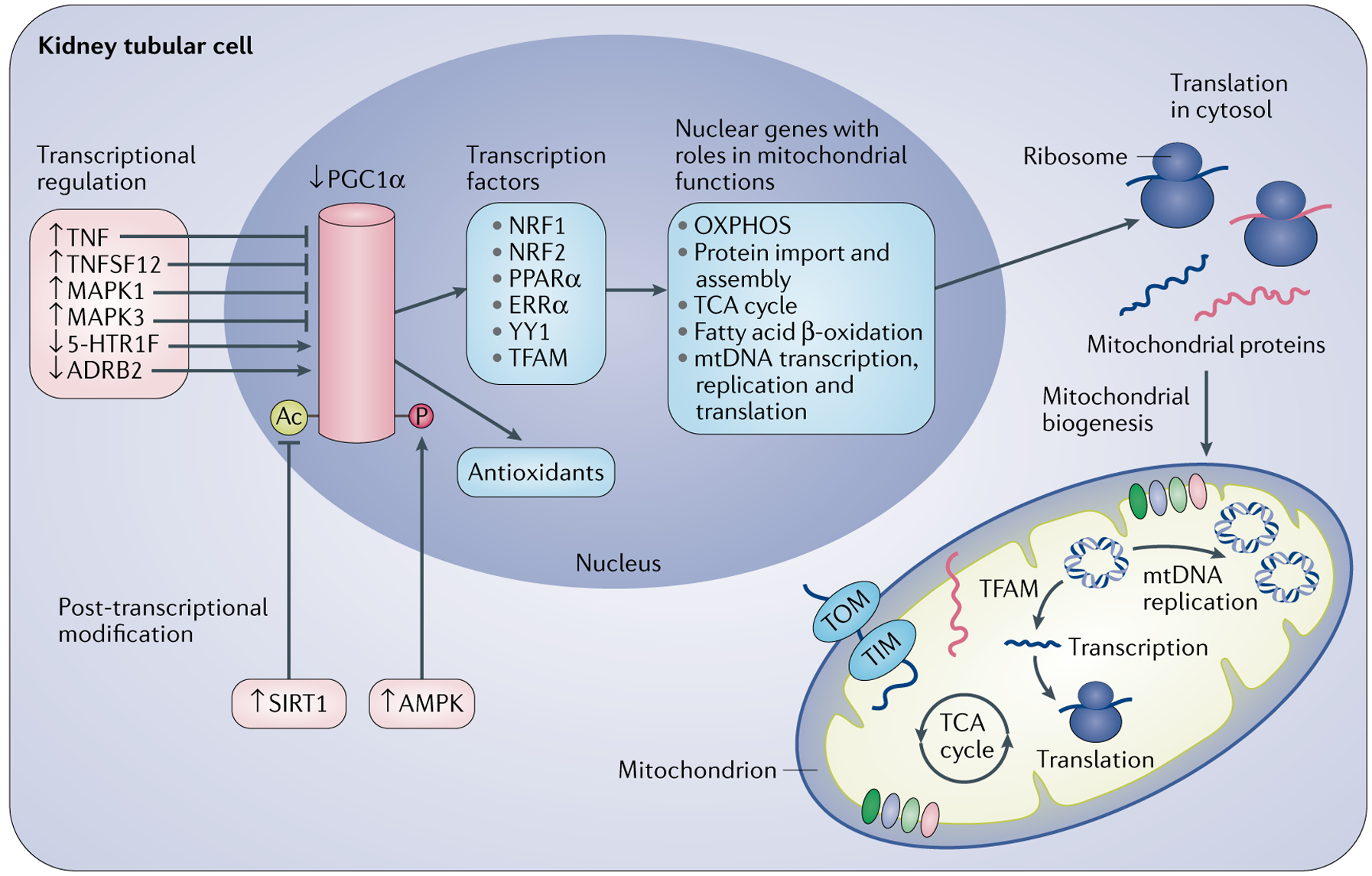
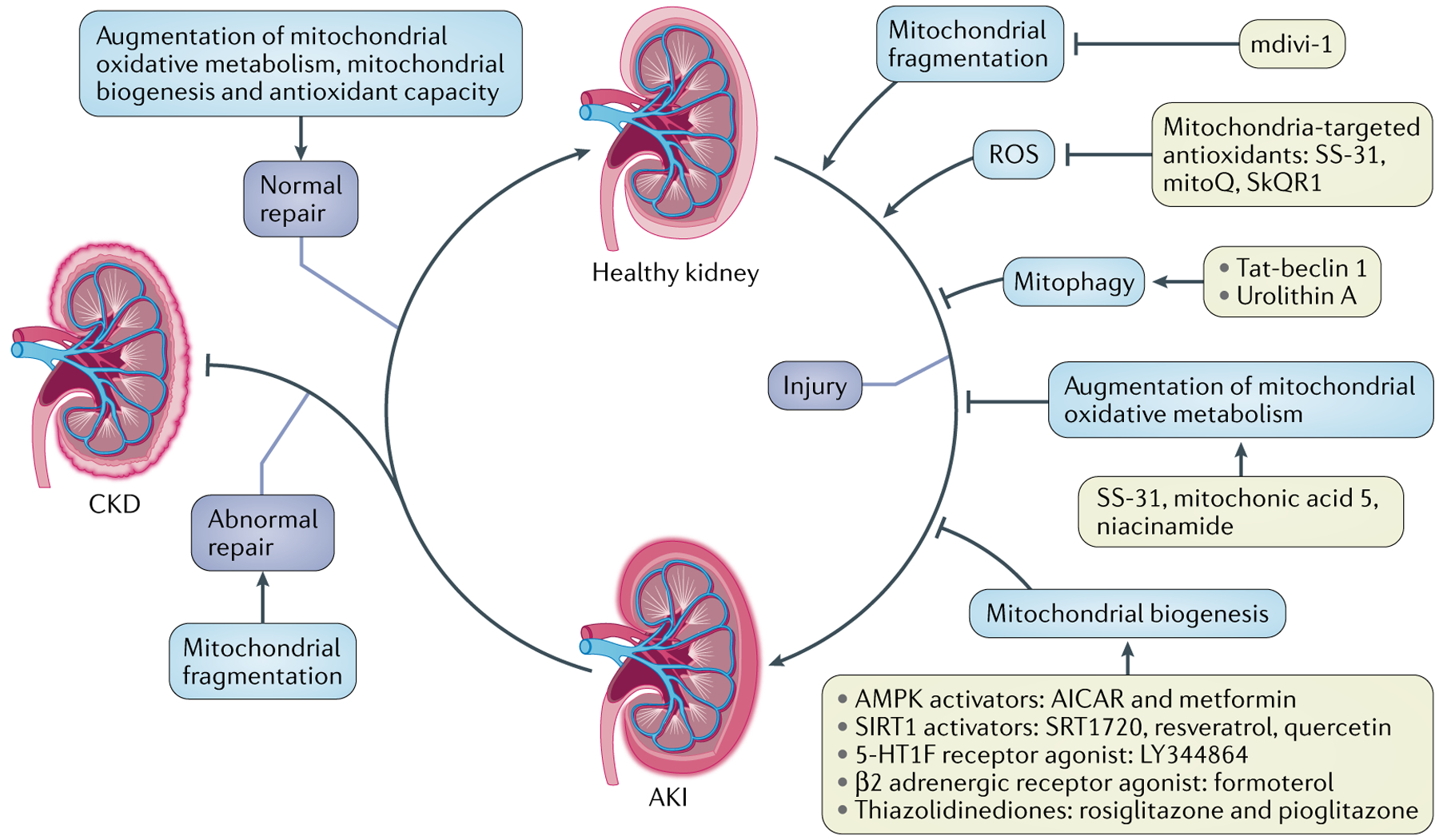
Mitochondria are essential for the activity, function and viability of eukaryotic cells and mitochondrial dysfunction is involved in the pathogenesis of acute kidney injury (AKI) and chronic kidney disease, as well as in abnormal kidney repair after AKI. Multiple quality control mechanisms, including antioxidant defence, protein quality control, mitochondrial DNA repair, mitochondrial dynamics, mitophagy and mitochondrial biogenesis, have evolved to preserve mitochondrial homeostasis under physiological and pathological conditions. Loss of these mechanisms may induce mitochondrial damage and dysfunction, leading to cell death, tissue injury and, potentially, organ failure. Accumulating evidence suggests a role of disturbances in mitochondrial quality control in the pathogenesis of AKI, incomplete or maladaptive kidney repair and chronic kidney disease. Moreover, specific interventions that target mitochondrial quality control mechanisms to preserve and restore mitochondrial function have emerged as promising therapeutic strategies to prevent and treat kidney injury and accelerate kidney repair. However, clinical translation of these findings is challenging owing to potential adverse effects, unclear mechanisms of action and a lack of knowledge of the specific roles and regulation of mitochondrial quality control mechanisms in kidney resident and circulating cell types during injury and repair of the kidney.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
-
- Guder WG & Ross BD Enzyme distribution along the nephron. Kidney Int. 26, 101–111 (1984). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
