

Temporal signal and the phylodynamic threshold of SARS-CoV-2
- PMID: 33235813
- PMCID: PMC7454936
- DOI: 10.1093/ve/veaa061
Temporal signal and the phylodynamic threshold of SARS-CoV-2
Abstract
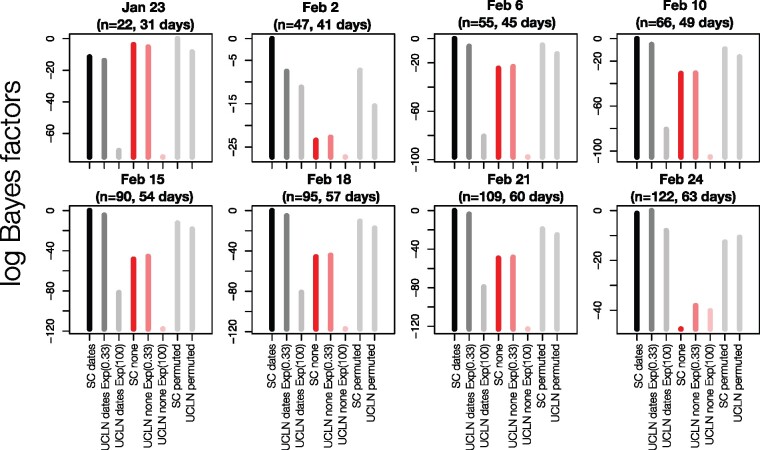
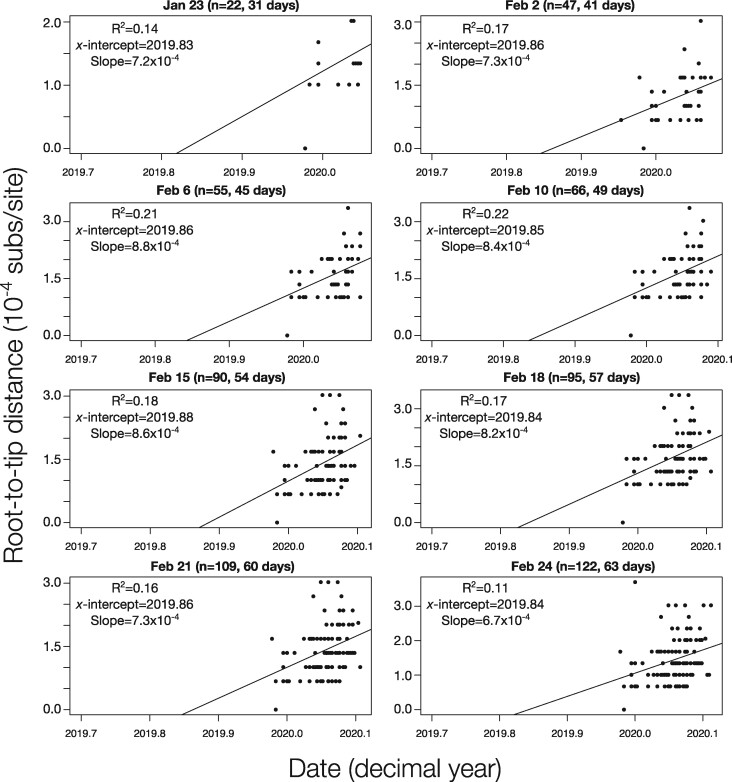
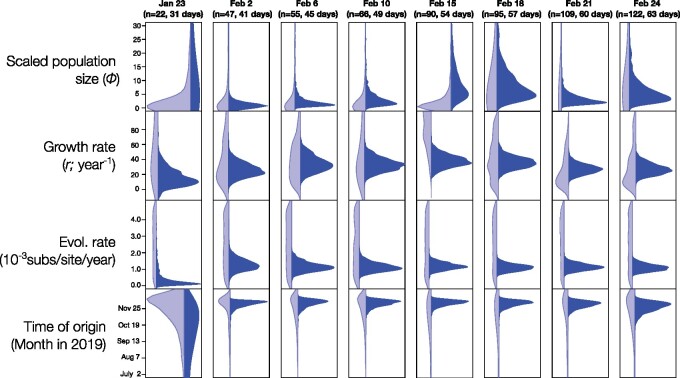
The ongoing SARS-CoV-2 outbreak marks the first time that large amounts of genome sequence data have been generated and made publicly available in near real time. Early analyses of these data revealed low sequence variation, a finding that is consistent with a recently emerging outbreak, but which raises the question of whether such data are sufficiently informative for phylogenetic inferences of evolutionary rates and time scales. The phylodynamic threshold is a key concept that refers to the point in time at which sufficient molecular evolutionary change has accumulated in available genome samples to obtain robust phylodynamic estimates. For example, before the phylodynamic threshold is reached, genomic variation is so low that even large amounts of genome sequences may be insufficient to estimate the virus's evolutionary rate and the time scale of an outbreak. We collected genome sequences of SARS-CoV-2 from public databases at eight different points in time and conducted a range of tests of temporal signal to determine if and when the phylodynamic threshold was reached, and the range of inferences that could be reliably drawn from these data. Our results indicate that by 2 February 2020, estimates of evolutionary rates and time scales had become possible. Analyses of subsequent data sets, that included between 47 and 122 genomes, converged at an evolutionary rate of about 1.1 × 10-3 subs/site/year and a time of origin of around late November 2019. Our study provides guidelines to assess the phylodynamic threshold and demonstrates that establishing this threshold constitutes a fundamental step for understanding the power and limitations of early data in outbreak genome surveillance.
Keywords: 2019 novel coronavirus (SARS-CoV-2); molecular clock; phylodynamic threshold; phylogenetics; severe acute respiratory syndrome corona virus 2; temporal signal.
© The Author(s) 2020. Published by Oxford University Press.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
