

CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations
- PMID: 33239111
- PMCID: PMC7690190
- DOI: 10.1186/s40478-020-01084-4
CGG expansion in NOTCH2NLC is associated with oculopharyngodistal myopathy with neurological manifestations
Abstract
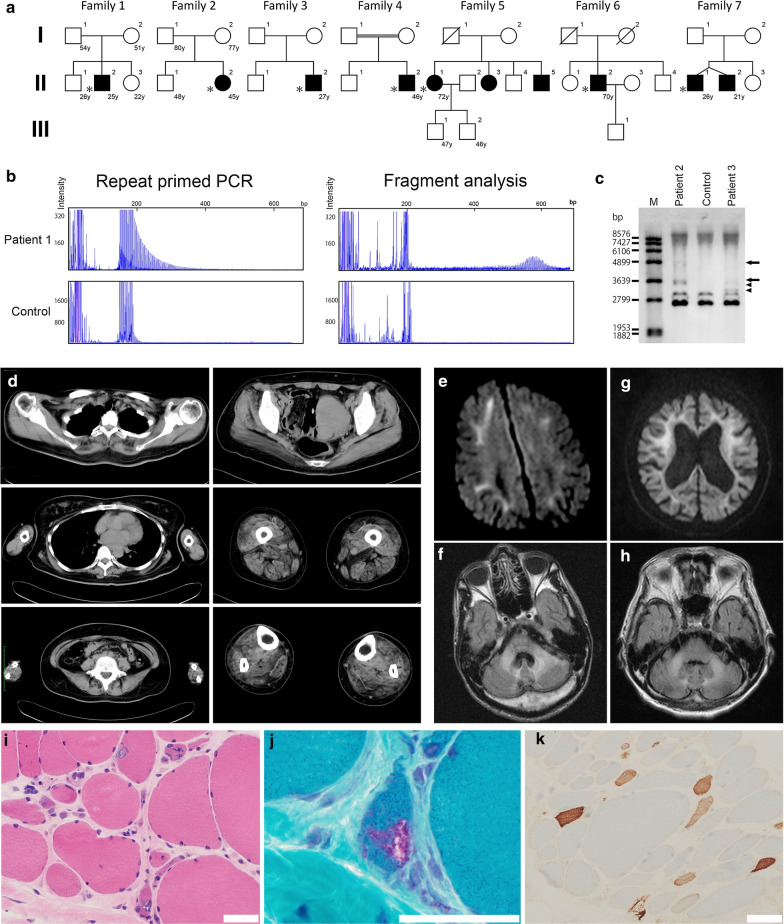
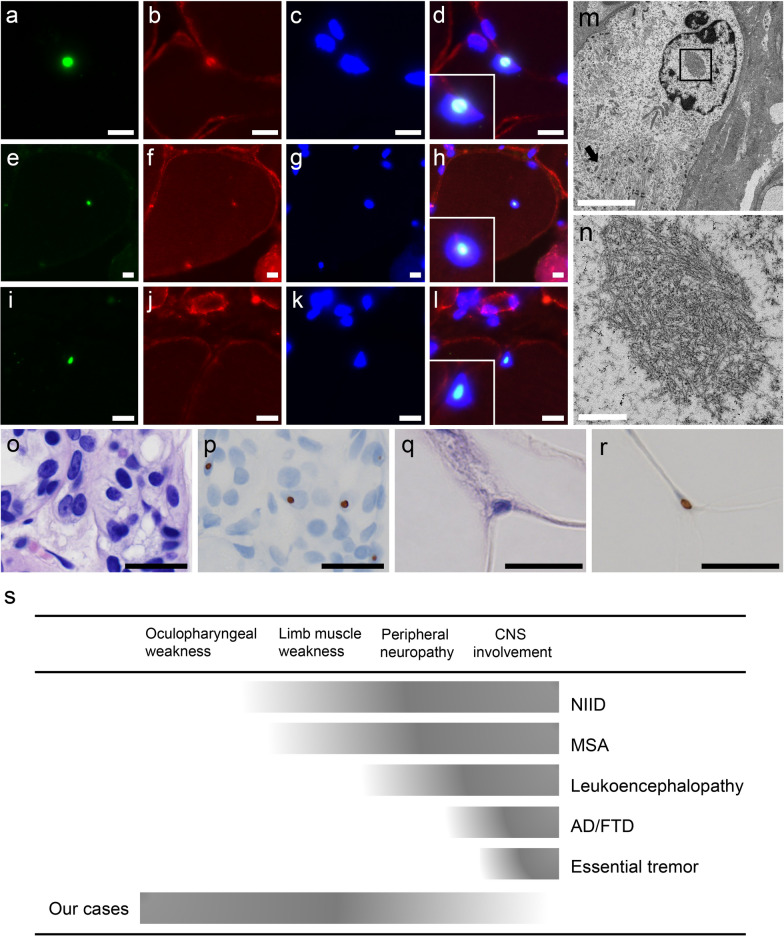
Oculopharyngodistal myopathy (OPDM) is a rare hereditary muscle disease characterized by progressive distal limb weakness, ptosis, ophthalmoplegia, bulbar muscle weakness and rimmed vacuoles on muscle biopsy. Recently, CGG repeat expansions in the noncoding regions of two genes, LRP12 and GIPC1, have been reported to be causative for OPDM. Furthermore, neuronal intranuclear inclusion disease (NIID) has been recently reported to be caused by CGG repeat expansions in NOTCH2NLC. We aimed to identify and to clinicopathologically characterize patients with OPDM who have CGG repeat expansions in NOTCH2NLC (OPDM_NOTCH2NLC). Note that 211 patients from 201 families, who were clinically or clinicopathologically diagnosed with OPDM or oculopharyngeal muscular dystrophy, were screened for CGG expansions in NOTCH2NLC by repeat primed-PCR. Clinical information and muscle pathology slides of identified patients with OPDM_NOTCH2NLC were re-reviewed. Intra-myonuclear inclusions were evaluated using immunohistochemistry and electron microscopy (EM). Seven Japanese OPDM patients had CGG repeat expansions in NOTCH2NLC. All seven patients clinically demonstrated ptosis, ophthalmoplegia, dysarthria and muscle weakness; they myopathologically had intra-myonuclear inclusions stained with anti-poly-ubiquitinated proteins, anti-SUMO1 and anti-p62 antibodies, which were diagnostic of NIID (typically on skin biopsy), in addition to rimmed vacuoles. The sample for EM was available only from one patient, which demonstrated intranuclear inclusions of 12.6 ± 1.6 nm in diameter. We identified seven patients with OPDM_NOTCH2NLC. Our patients had various additional central and/or peripheral nervous system involvement, although all were clinicopathologically compatible; thus, they were diagnosed as having OPDM and expanding a phenotype of the neuromyodegenerative disease caused by CGG repeat expansions in NOTCH2NLC.
Keywords: CGG repeat expansion; NOTCH2NLC; Neuronal intranuclear inclusion disease; Oculopharyngeal muscular dystrophy; Oculopharyngodistal myopathy.
Conflict of interest statement
The authors report no competing interests.
Figures
References
-
- Hayashi T, Katagiri S, Mizobuchi K, Yoshitake K, Kameya S, Matsuura T, Iwata T, Nakano T. Heterozygous GGC repeat expansion of NOTCH2NLC in a patient with neuronal intranuclear inclusion disease and progressive retinal dystrophy. Ophthal Genet. 2020;41:93–95. doi: 10.1080/13816810.2020.1723119. - DOI - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
