

Fibrosis: from mechanisms to medicines
- PMID: 33239795
- PMCID: PMC8034822
- DOI: 10.1038/s41586-020-2938-9
Fibrosis: from mechanisms to medicines
Abstract
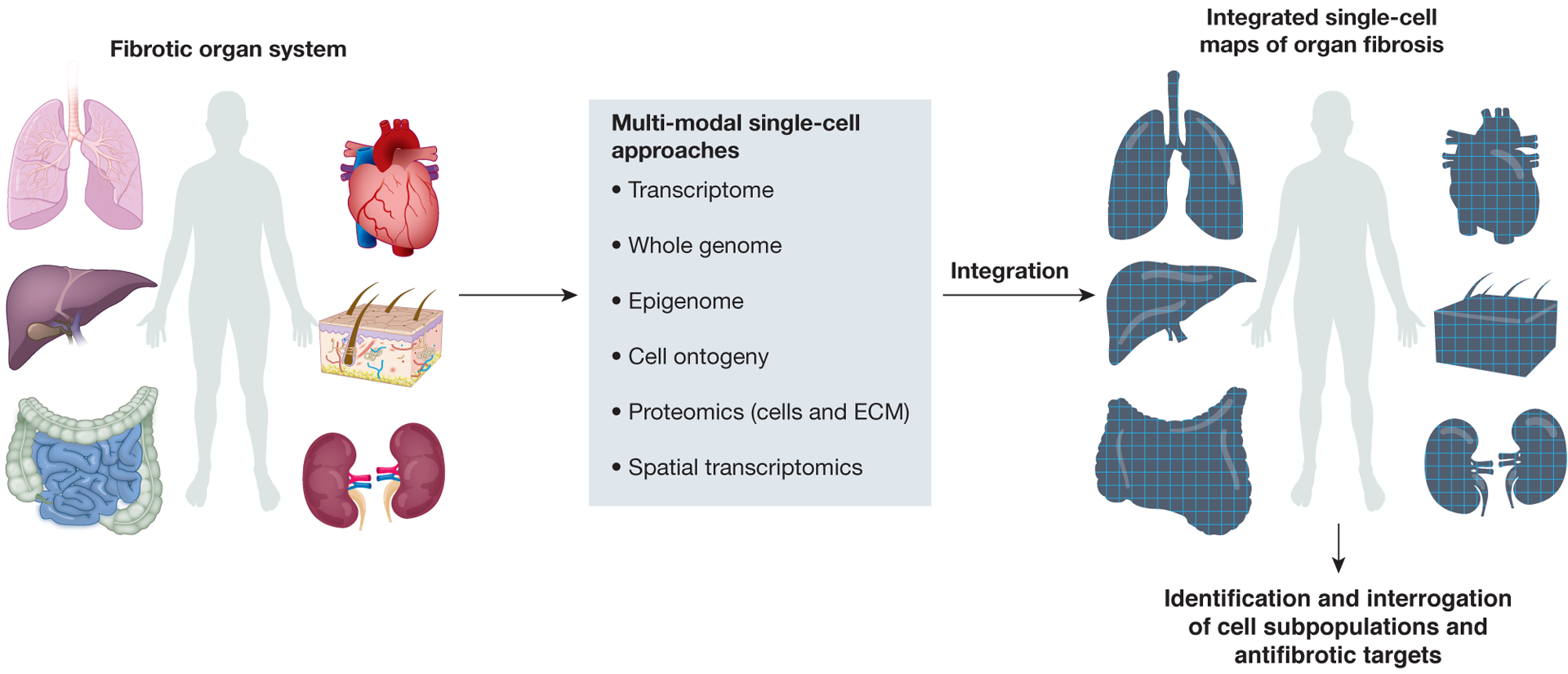
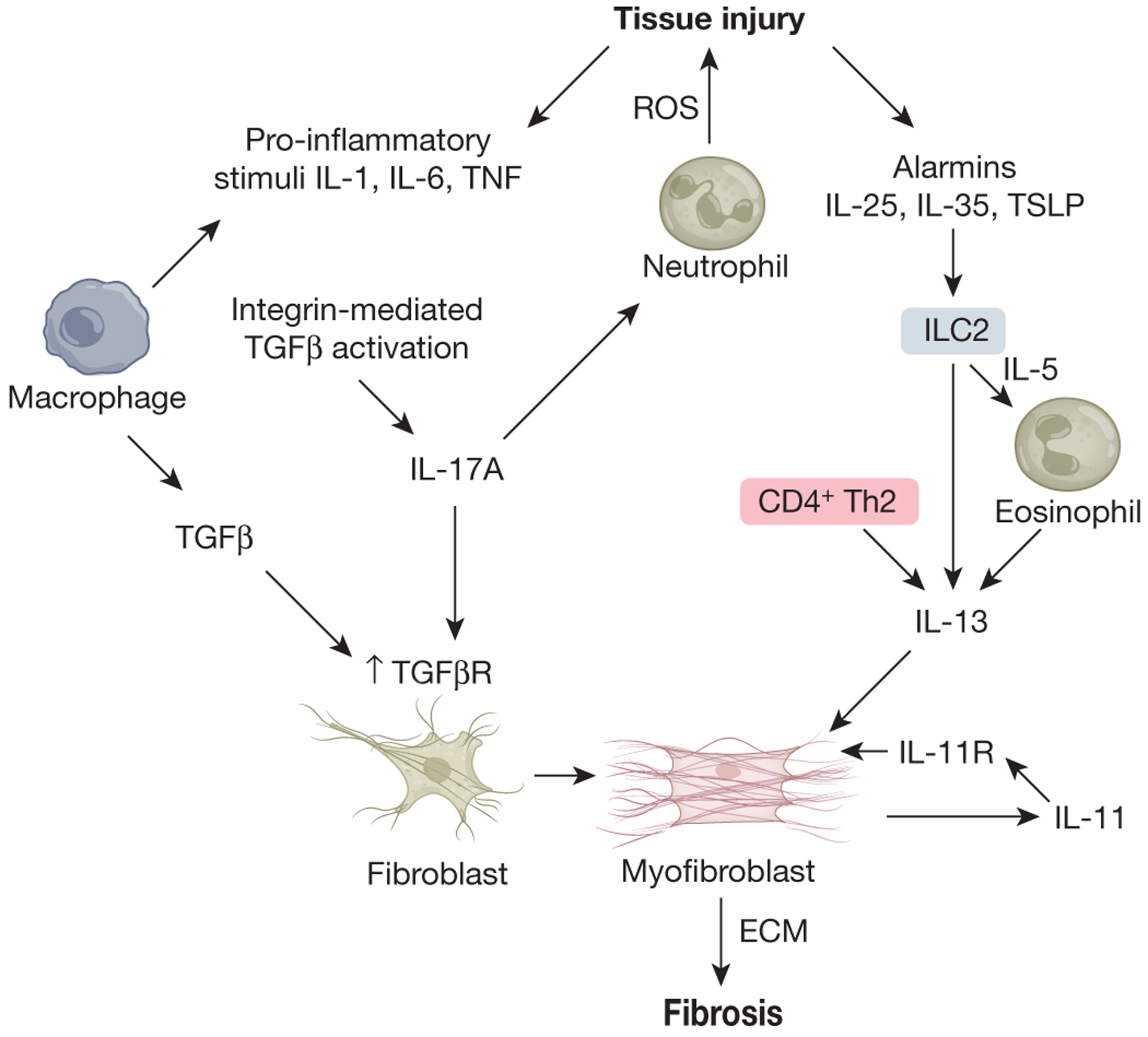
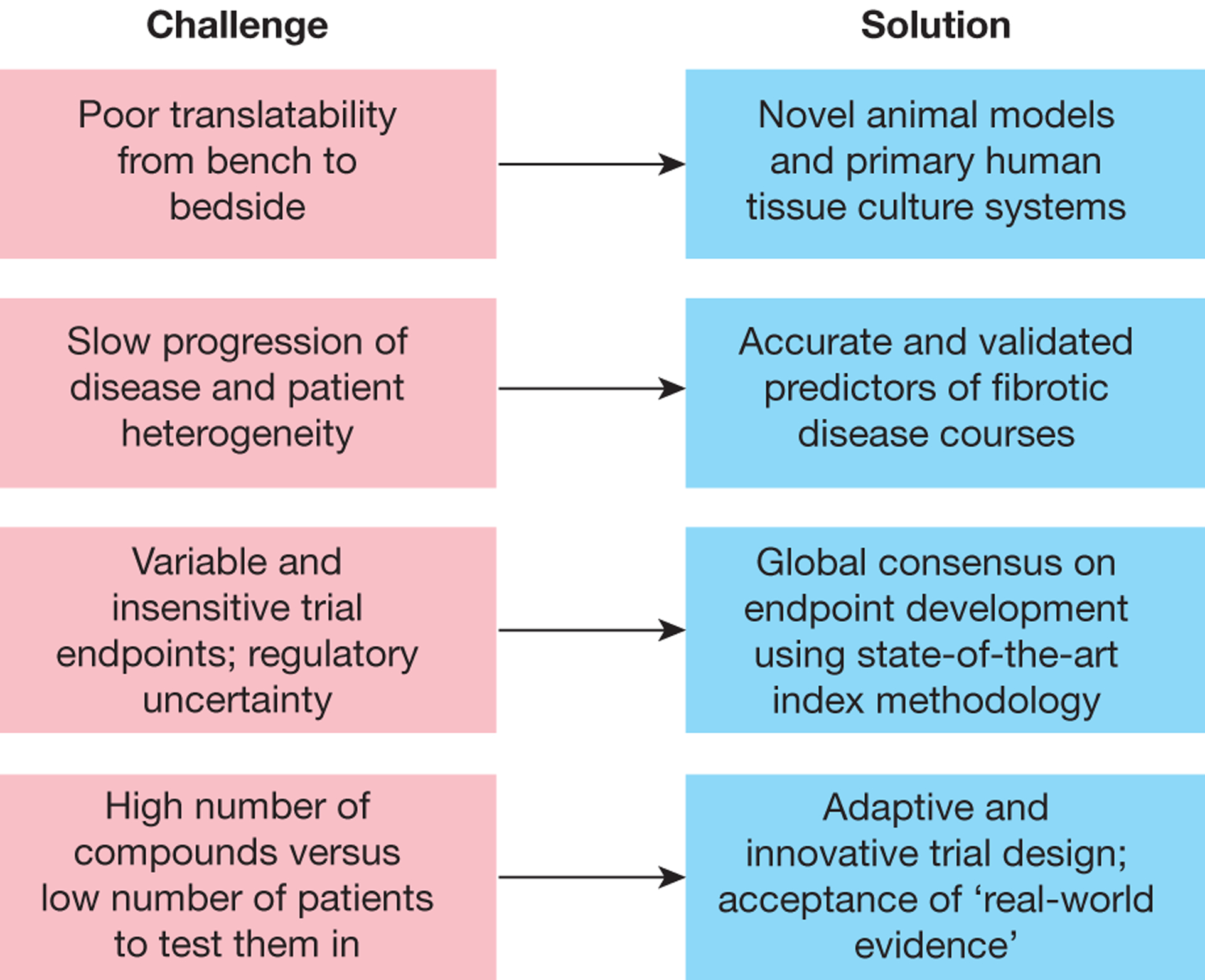
Fibrosis can affect any organ and is responsible for up to 45% of all deaths in the industrialized world. It has long been thought to be relentlessly progressive and irreversible, but both preclinical models and clinical trials in various organ systems have shown that fibrosis is a highly dynamic process. This has clear implications for therapeutic interventions that are designed to capitalize on this inherent plasticity. However, despite substantial progress in our understanding of the pathobiology of fibrosis, a translational gap remains between the identification of putative antifibrotic targets and conversion of this knowledge into effective treatments in humans. Here we discuss the transformative experimental strategies that are being leveraged to dissect the key cellular and molecular mechanisms that regulate fibrosis, and the translational approaches that are enabling the emergence of precision medicine-based therapies for patients with fibrosis.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
