

Higher Incidence of B Cell Malignancies in Primary Immunodeficiencies: A Combination of Intrinsic Genomic Instability and Exocytosis Defects at the Immunological Synapse
- PMID: 33240268
- PMCID: PMC7680899
- DOI: 10.3389/fimmu.2020.581119
Higher Incidence of B Cell Malignancies in Primary Immunodeficiencies: A Combination of Intrinsic Genomic Instability and Exocytosis Defects at the Immunological Synapse
Abstract
Congenital defects of the immune system called primary immunodeficiency disorders (PID) describe a group of diseases characterized by a decrease, an absence, or a malfunction of at least one part of the immune system. As a result, PID patients are more prone to develop life-threatening complications, including cancer. PID currently include over 400 different disorders, however, the variety of PID-related cancers is narrow. We discuss here reasons for this clinical phenotype. Namely, PID can lead to cell intrinsic failure to control cell transformation, failure to activate tumor surveillance by cytotoxic cells or both. As the most frequent tumors seen among PID patients stem from faulty lymphocyte development leading to leukemia and lymphoma, we focus on the extensive genomic alterations needed to create the vast diversity of B and T lymphocytes with potential to recognize any pathogen and why defects in these processes lead to malignancies in the immunodeficient environment of PID patients. In the second part of the review, we discuss PID affecting tumor surveillance and especially membrane trafficking defects caused by altered exocytosis and regulation of the actin cytoskeleton. As an impairment of these membrane trafficking pathways often results in dysfunctional effector immune cells, tumor cell immune evasion is elevated in PID. By considering new anti-cancer treatment concepts, such as transfer of genetically engineered immune cells, restoration of anti-tumor immunity in PID patients could be an approach to complement standard therapies.
Keywords: B cells; actin cytoskeleton; cancer; cytotoxic cells; exocytosis; immunological synapse; membrane trafficking; primary immunodeficiencies.
Copyright © 2020 Mastio, Saeed, Wurzer, Krecke, Westerberg and Thomas.
Figures
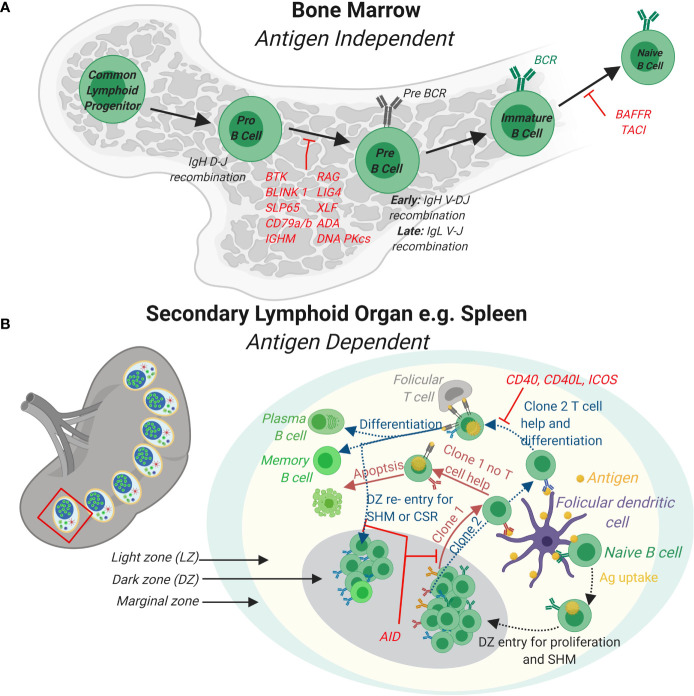
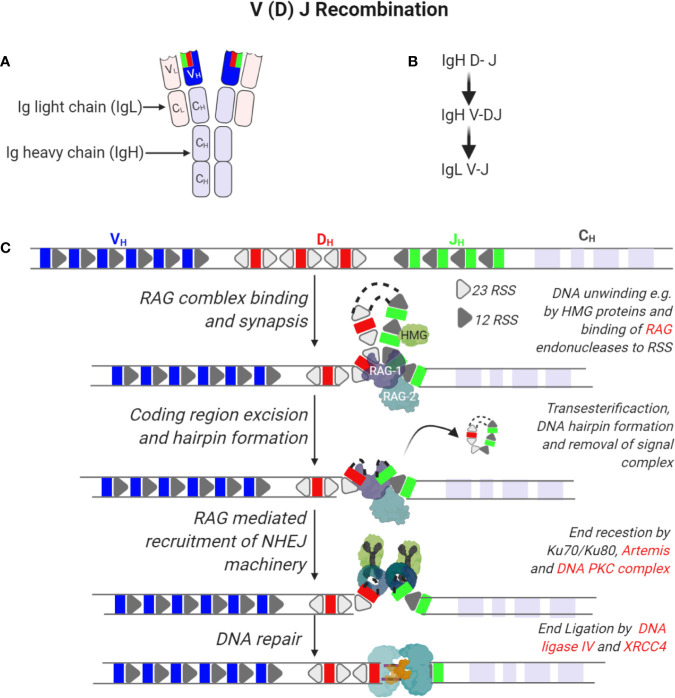
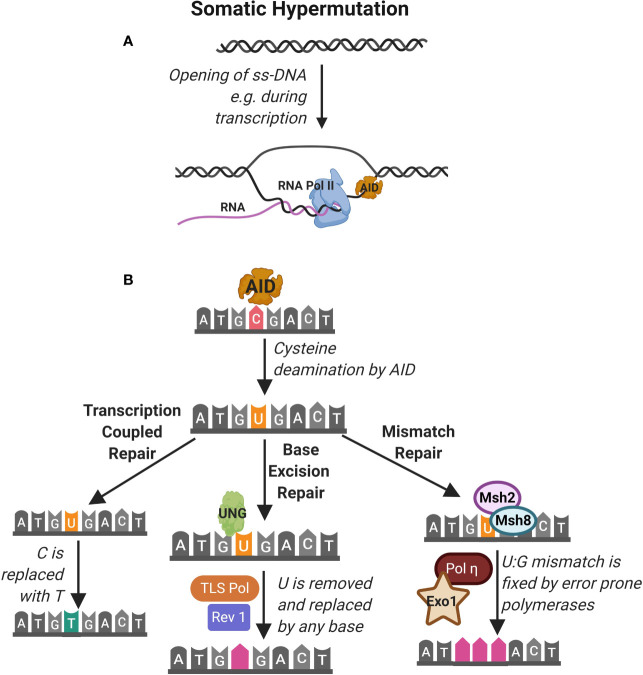
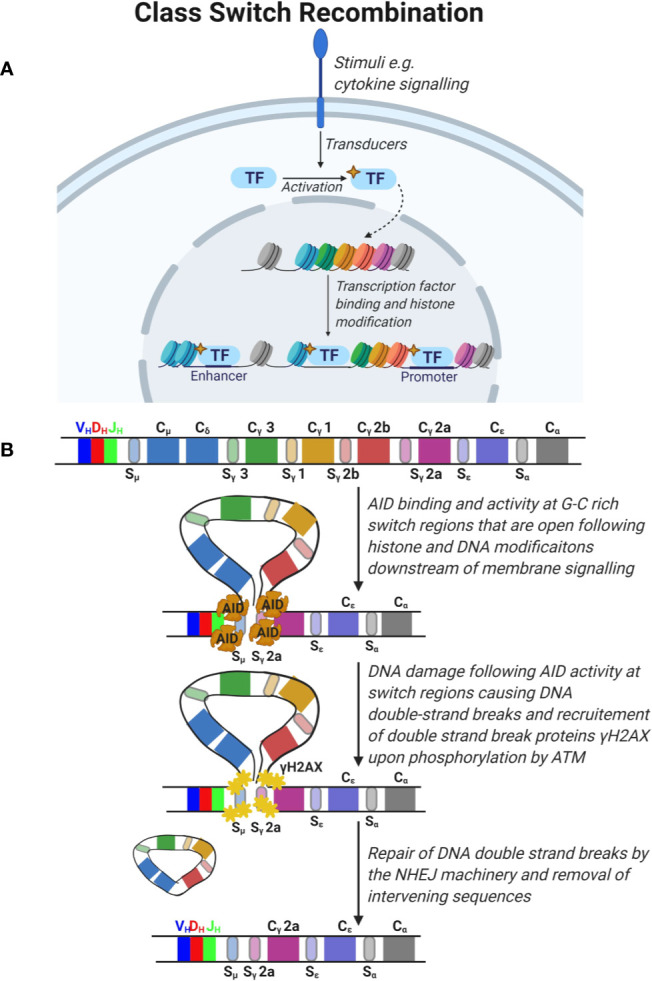
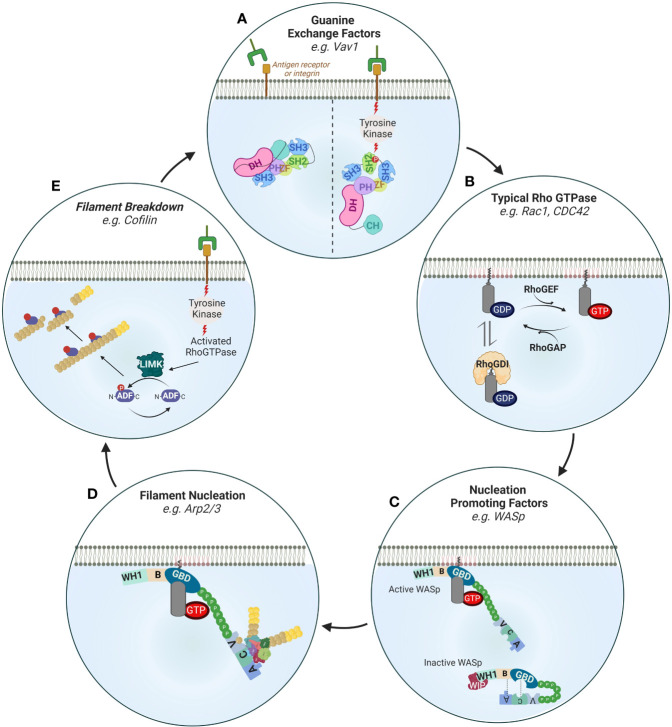
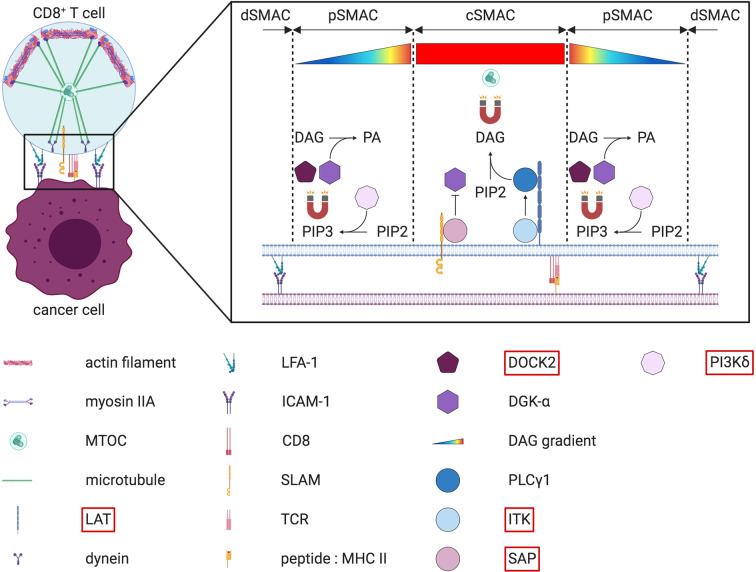
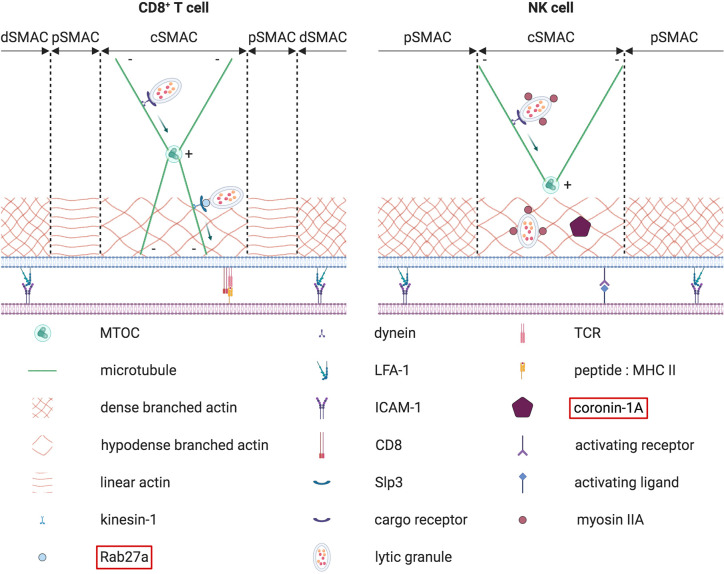
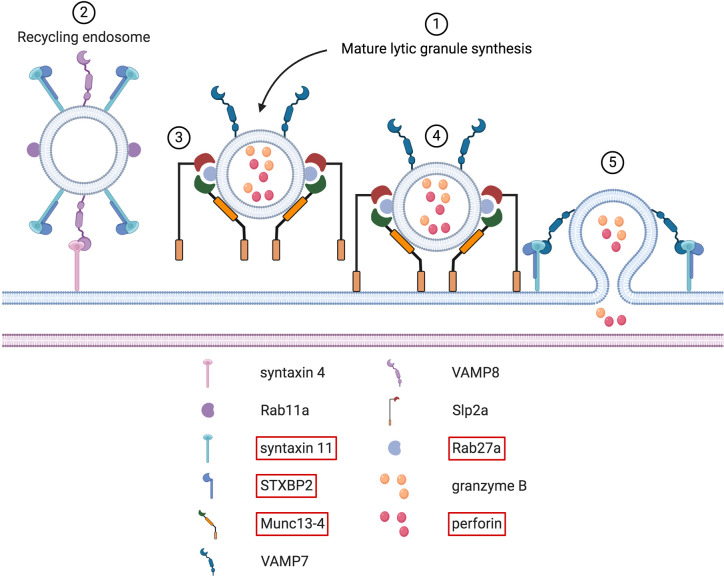
References
-
- Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update on the Classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol (2020) 40(1):24–64. 10.1007/s10875-019-00737-x - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials