

Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis
- PMID: 33242422
- PMCID: PMC7864894
- DOI: 10.1016/j.neuron.2020.11.005
Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis
Abstract
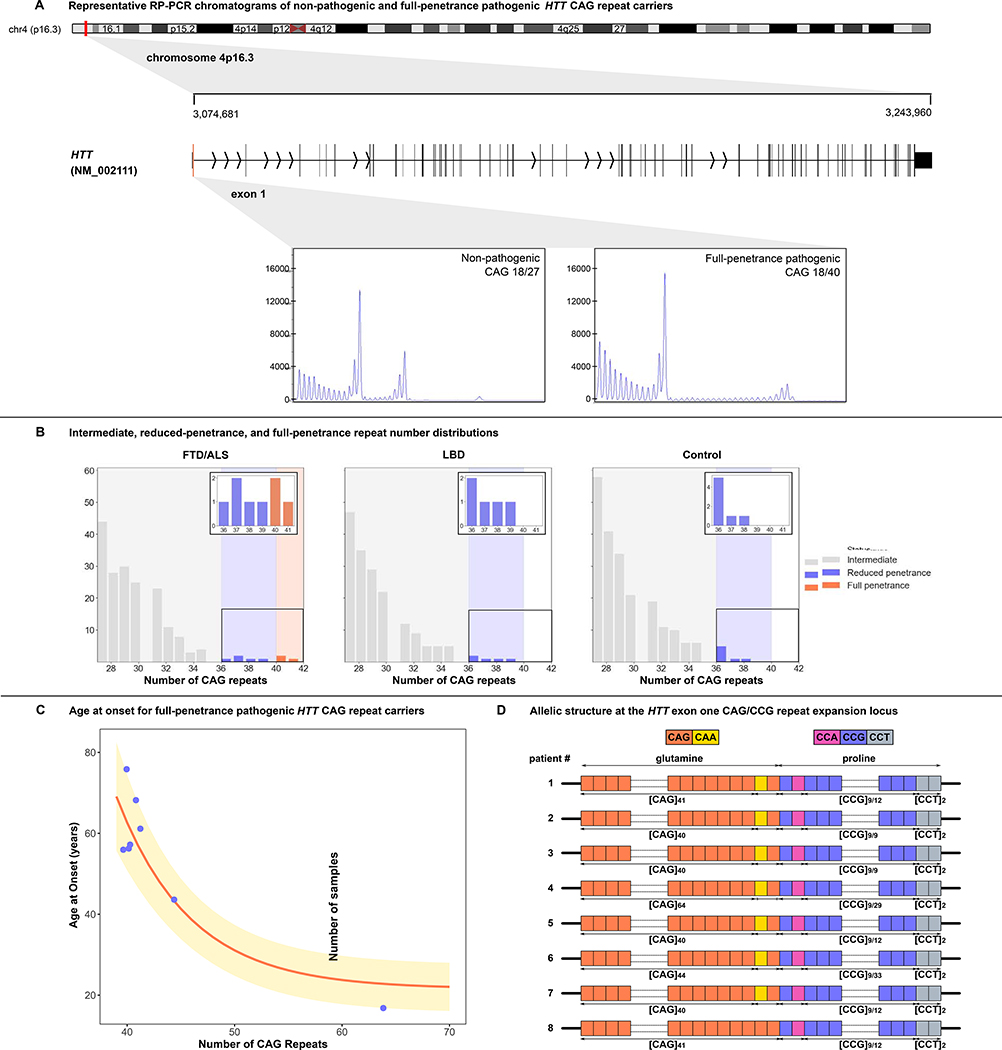
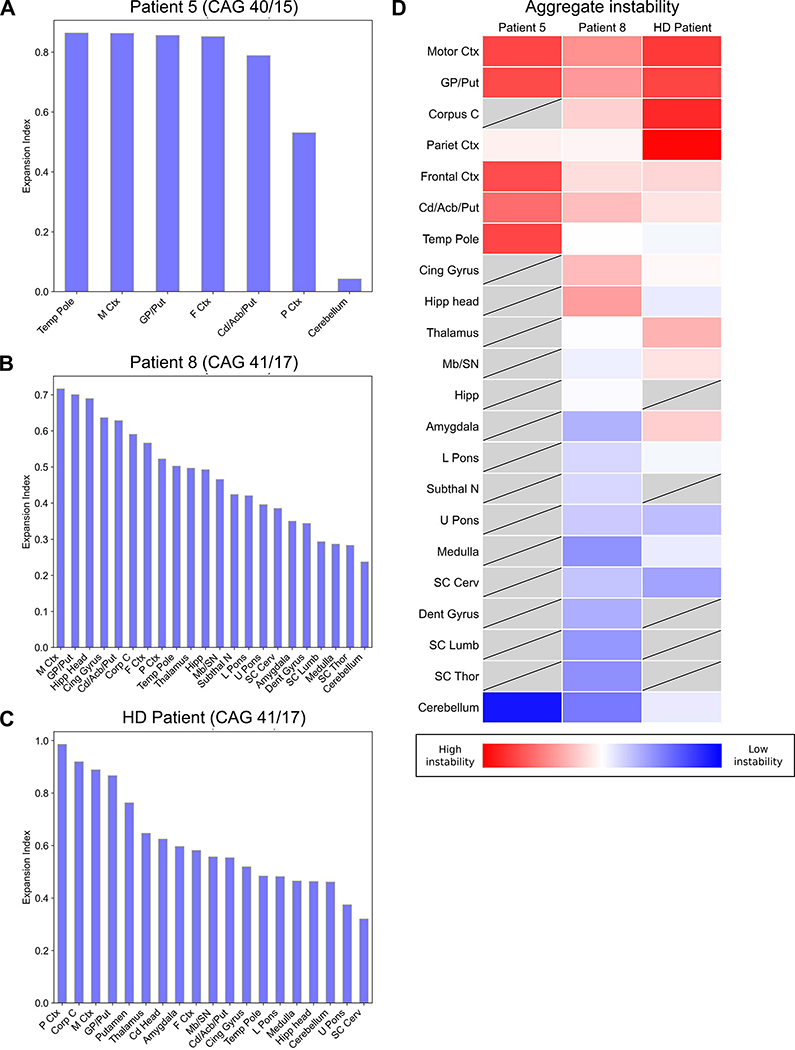
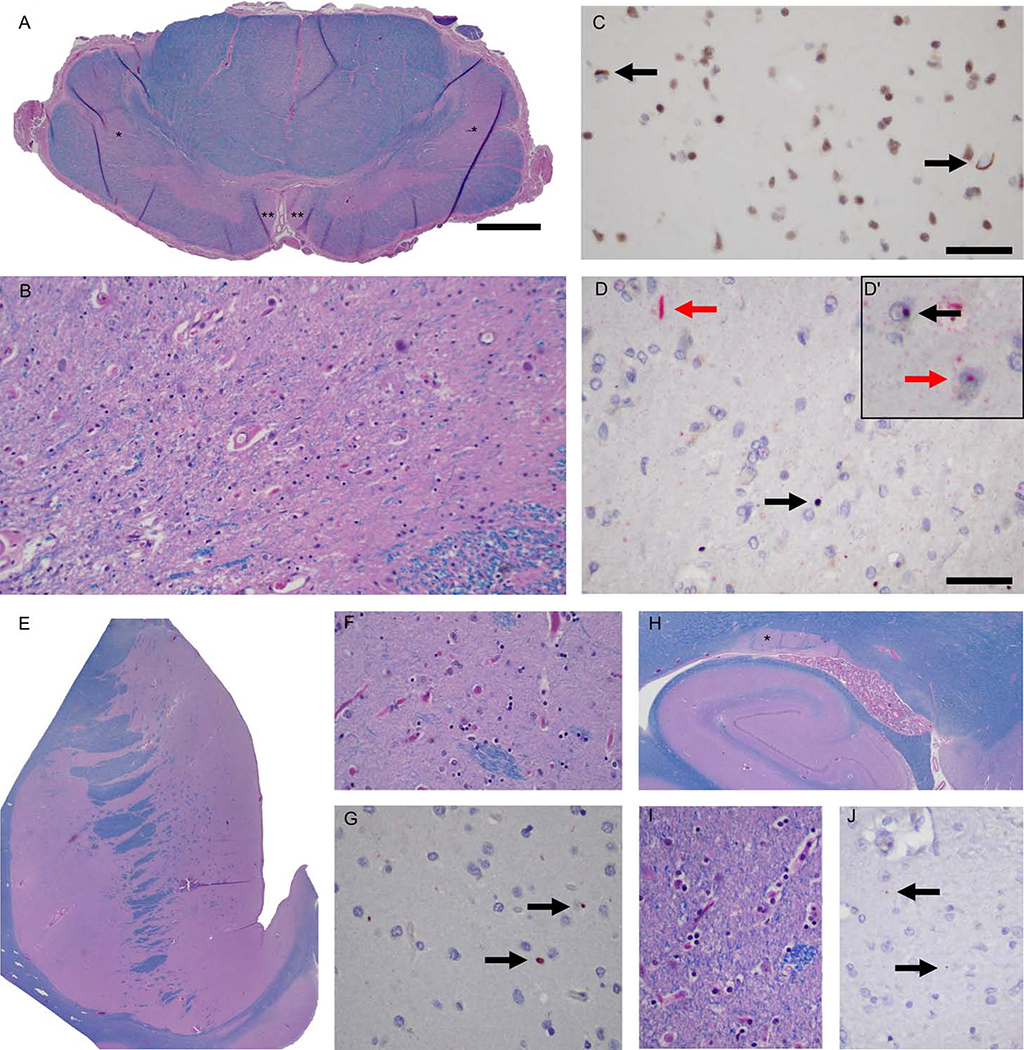
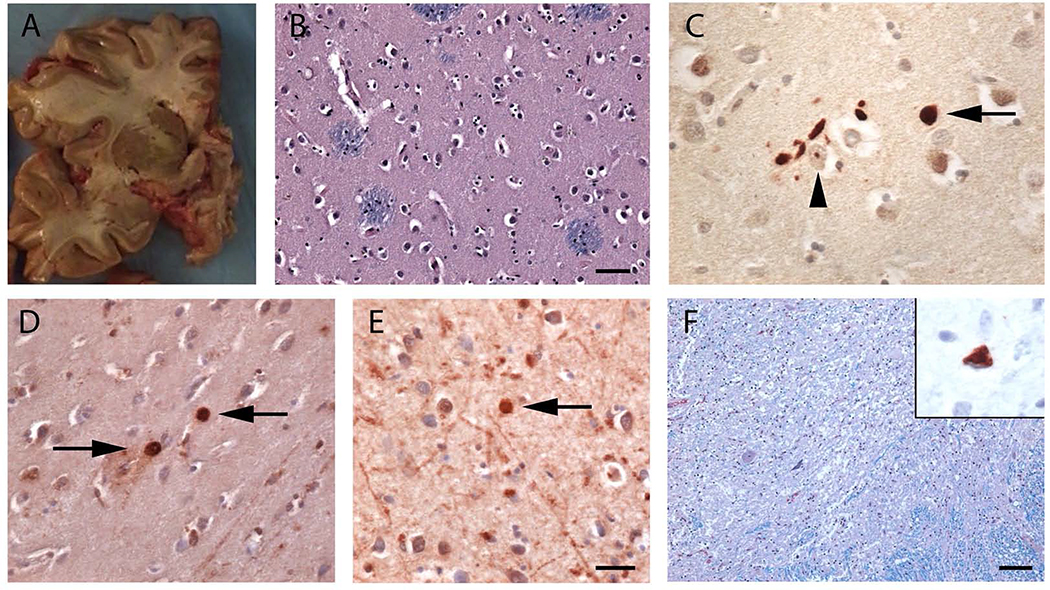
We examined the role of repeat expansions in the pathogenesis of frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) by analyzing whole-genome sequence data from 2,442 FTD/ALS patients, 2,599 Lewy body dementia (LBD) patients, and 3,158 neurologically healthy subjects. Pathogenic expansions (range, 40-64 CAG repeats) in the huntingtin (HTT) gene were found in three (0.12%) patients diagnosed with pure FTD/ALS syndromes but were not present in the LBD or healthy cohorts. We replicated our findings in an independent collection of 3,674 FTD/ALS patients. Postmortem evaluations of two patients revealed the classical TDP-43 pathology of FTD/ALS, as well as huntingtin-positive, ubiquitin-positive aggregates in the frontal cortex. The neostriatal atrophy that pathologically defines Huntington's disease was absent in both cases. Our findings reveal an etiological relationship between HTT repeat expansions and FTD/ALS syndromes and indicate that genetic screening of FTD/ALS patients for HTT repeat expansions should be considered.
Keywords: amyotrophic lateral sclerosis; frontotemporal dementia; huntingtin; repeat expansions; whole-genome sequencing.
Published by Elsevier Inc.
Conflict of interest statement
Declaration of Interests S.P.-B., A.B.S., J.A.H., H.R.M., and B.J.T. hold US, EU, and Canadian patents on the clinical testing and therapeutic intervention for the hexanucleotide repeat expansion of C9 or f72. S.W.S. serves on the scientific advisory council of the Lewy Body Dementia Association and is an editorial board member for the Journal of Parkinson’s Disease. B.J.T. is an editorial board member for JAMA Neurology, JNNP, and Neurobiology of Aging. V.S. is on the journal editorial boards of Amyotrophic Lateral Sclerosis, European Neurology, American Journal of Neurodegenerative Diseases, and Frontiers in Neurology. He has also received compensation for consulting services and speaking activities from AveXis, Cytokinetics, Italfarmaco, and Zambon. J.B.R. is an editor for Brain and has received compensation for consulting services from Asceneuron, Biogen, UCB, Astex, and SV Health. J.E.L. is a member of the scientific advisory board for Cerevel Therapeutics and a consultant and provides expert testimony for Perkins Coie.
Figures
Comment in
-
Questioning the causality of HTT CAG-repeat expansions in FTD/ALS.Neuron. 2021 Jun 16;109(12):1945-1946. doi: 10.1016/j.neuron.2021.04.010. Neuron. 2021. PMID: 34139183 No abstract available.
-
Highlighting the clinical potential of HTT repeat expansions in Frontotemporal Dementia and Amyotrophic Lateral Sclerosis.Neuron. 2021 Jun 16;109(12):1947-1948. doi: 10.1016/j.neuron.2021.04.020. Neuron. 2021. PMID: 34139184 No abstract available.
References
-
- Bean L, and Bayrak-Toydemir P (2014). American College of Medical Genetics and Genomics Standards and Guidelines for Clinical Genetics Laboratories, 2014 edition: technical standards and guidelines for Huntington disease. Genet. Med 16, e2–e2. - PubMed
-
- Brooks BR (1994). El Escorial World Federation of Neurology criteria for the diagnosis of amyotrophic lateral sclerosis. Subcommittee on Motor Neuron Diseases/Amyotrophic Lateral Sclerosis of the World Federation of Neurology Research Group on Neuromuscular Diseases and the El Escorial “Clinical limits of amyotrophic lateral sclerosis” workshop contributors. J. Neurol. Sci 124 Suppl, 96–107. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 AG065463/AG/NIA NIH HHS/United States
- G0600974/MRC_/Medical Research Council/United Kingdom
- DH_/Department of Health/United Kingdom
- K-1501/PUK_/Parkinson's UK/United Kingdom
- ZIA NS003034/ImNIH/Intramural NIH HHS/United States
- J-0901/PUK_/Parkinson's UK/United Kingdom
- MR/L016397/1/MRC_/Medical Research Council/United Kingdom
- MC_UU_00005/12/MRC_/Medical Research Council/United Kingdom
- R01 NS115144/NS/NINDS NIH HHS/United States
- P30 AG013854/AG/NIA NIH HHS/United States
- MR/R011354/1/MRC_/Medical Research Council/United Kingdom
- P30 AG066507/AG/NIA NIH HHS/United States
- MR/S000992/1/MRC_/Medical Research Council/United Kingdom
- R56 NS073873/NS/NINDS NIH HHS/United States
- 103838/WT_/Wellcome Trust/United Kingdom
- ZIA AG000934/ImNIH/Intramural NIH HHS/United States
- MR/L501529/1/MRC_/Medical Research Council/United Kingdom
- I01 BX002466/BX/BLRD VA/United States
- MC_U105597119/MRC_/Medical Research Council/United Kingdom
- AL-CHALABI/APR15/844-791/MNDA_/Motor Neurone Disease Association/United Kingdom
- MC_PC_14089/MRC_/Medical Research Council/United Kingdom
- I01 BX005161/BX/BLRD VA/United States
- MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- ZIA AG000935/ImNIH/Intramural NIH HHS/United States
- R01 NS073873/NS/NINDS NIH HHS/United States
- MCLAUGHLIN/OCT15/957-799/MNDA_/Motor Neurone Disease Association/United Kingdom
- TURNER/OCT18/989-797/MNDA_/Motor Neurone Disease Association/United Kingdom
- P30 AG066462/AG/NIA NIH HHS/United States
- MR/T018569/1/MRC_/Medical Research Council/United Kingdom
- P01 AG066597/AG/NIA NIH HHS/United States
- MC_EX_MR/M009203/1/MRC_/Medical Research Council/United Kingdom
- G0400074/MRC_/Medical Research Council/United Kingdom
- MR/R024804/1/MRC_/Medical Research Council/United Kingdom
- ZIA NS003154/ImNIH/Intramural NIH HHS/United States
- WT_/Wellcome Trust/United Kingdom
- MR/S006753/1/MRC_/Medical Research Council/United Kingdom
- MC_EX_MR/N50192X/1/MRC_/Medical Research Council/United Kingdom
- MR/N026004/1/MRC_/Medical Research Council/United Kingdom
- P30 AG066514/AG/NIA NIH HHS/United States
- MR/M024962/1/MRC_/Medical Research Council/United Kingdom
- MR/N008324/1/MRC_/Medical Research Council/United Kingdom
- ZIA AG000933/ImNIH/Intramural NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
