

Enhancing calmodulin binding to cardiac ryanodine receptor completely inhibits pressure-overload induced hypertrophic signaling
- PMID: 33244105
- PMCID: PMC7691336
- DOI: 10.1038/s42003-020-01443-w
Enhancing calmodulin binding to cardiac ryanodine receptor completely inhibits pressure-overload induced hypertrophic signaling
Abstract
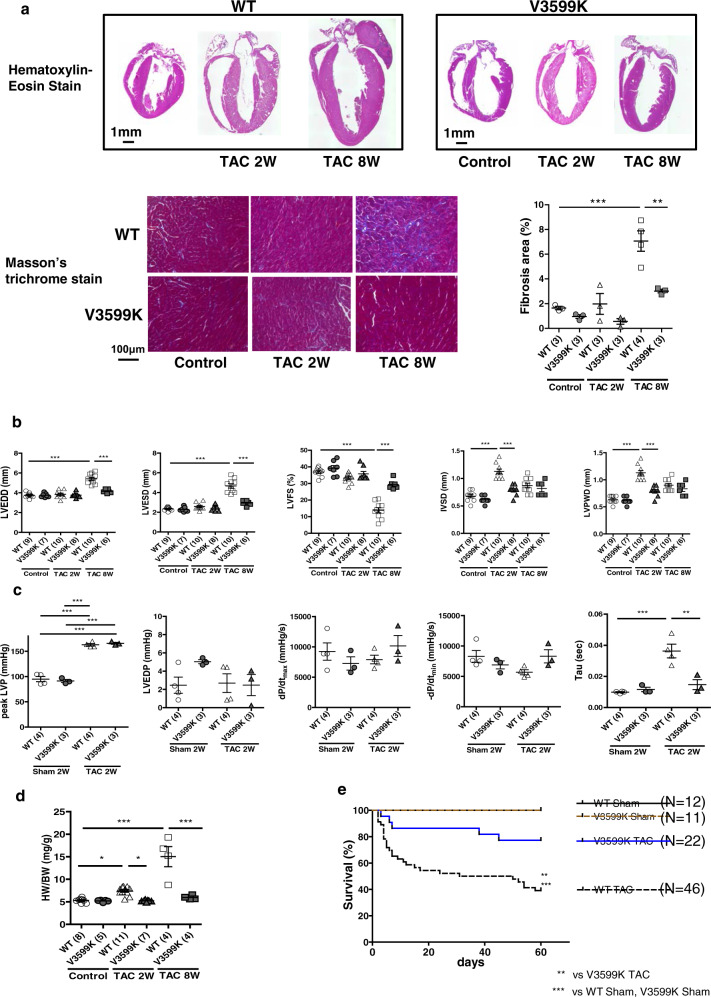
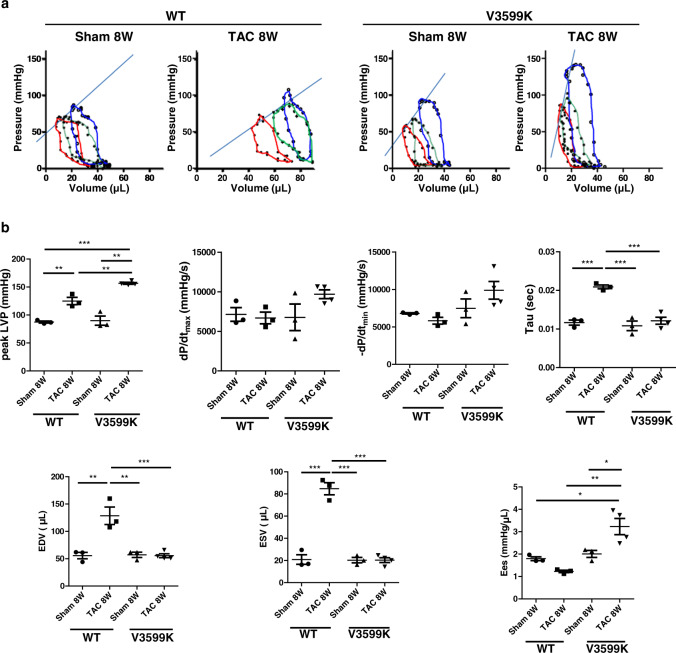
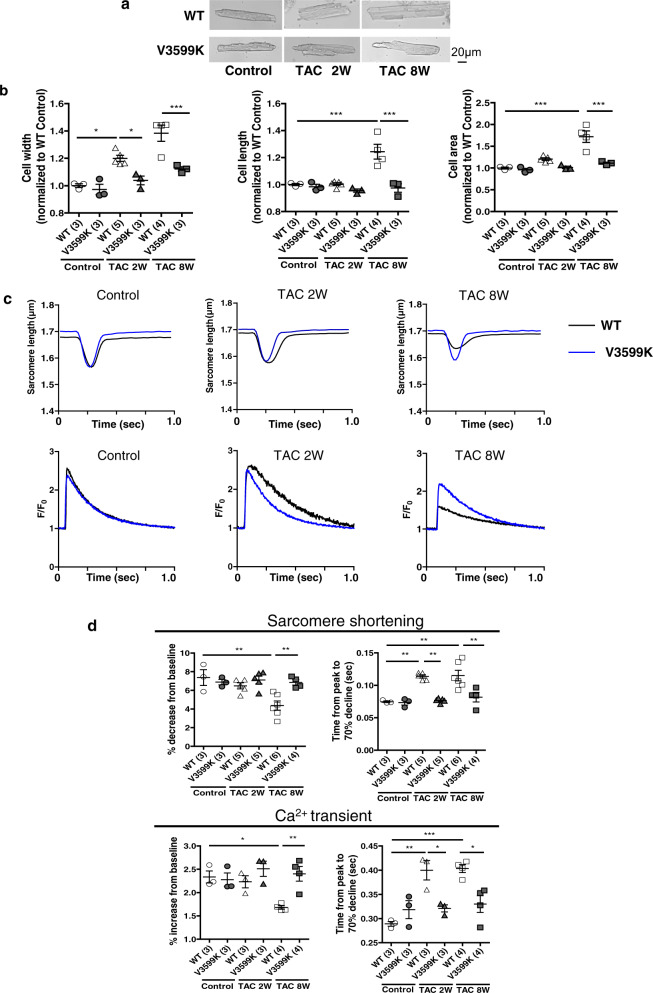
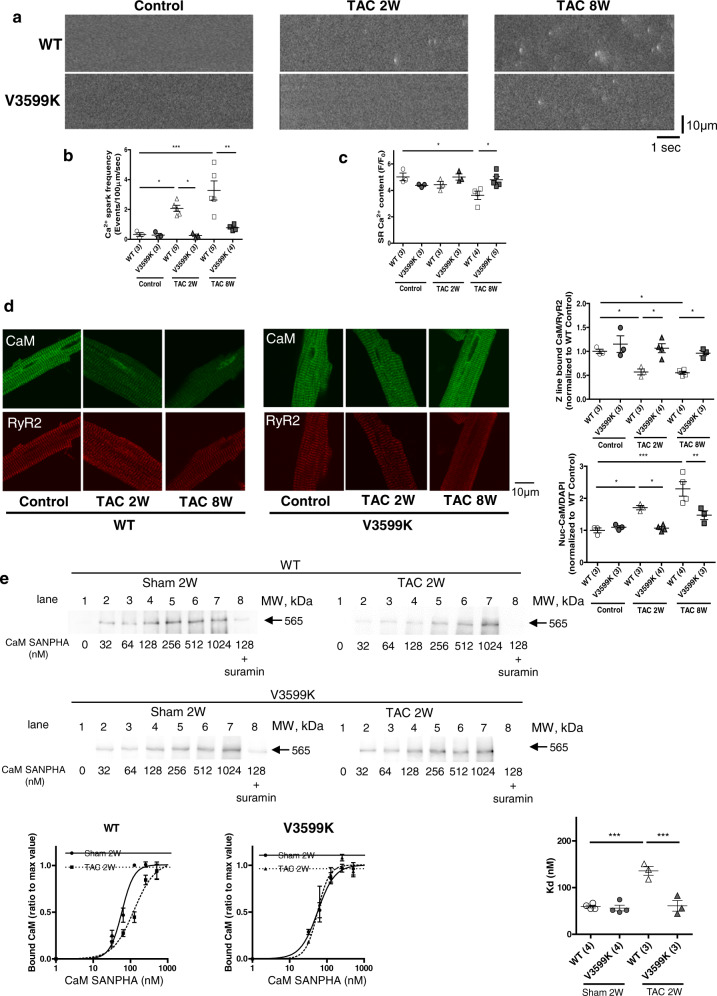
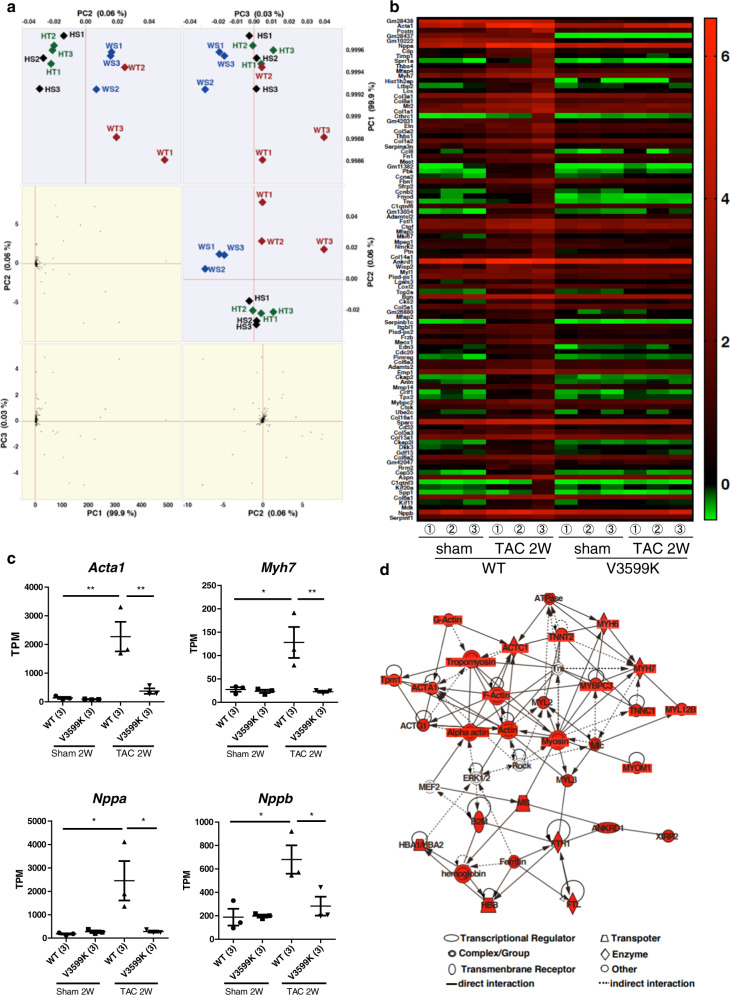
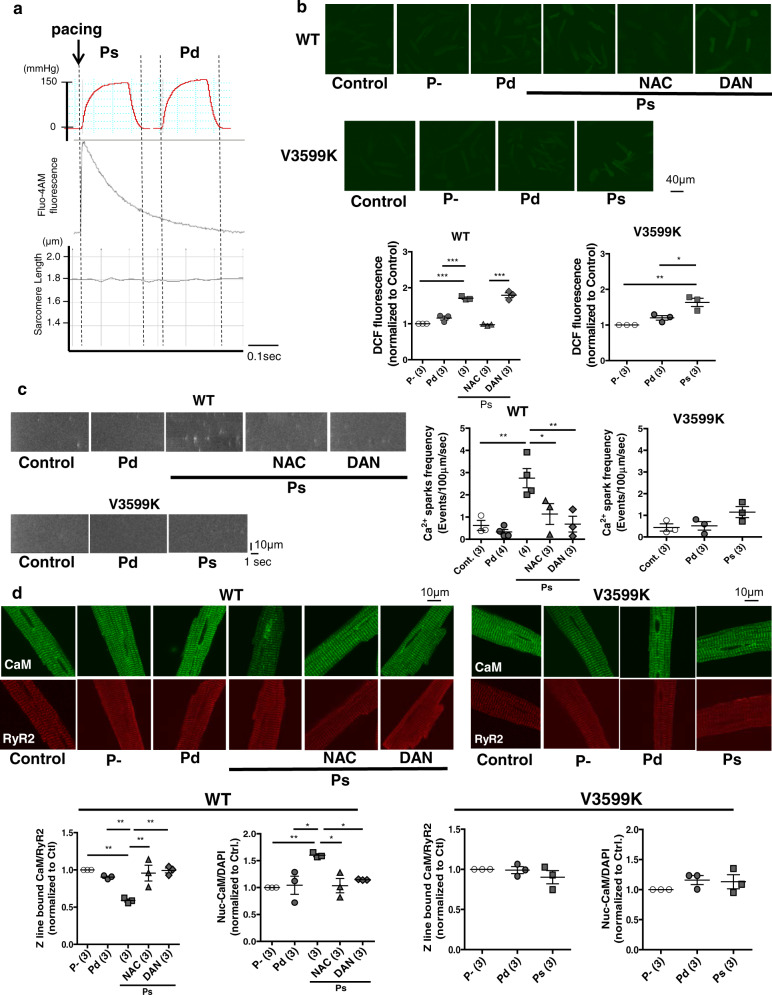
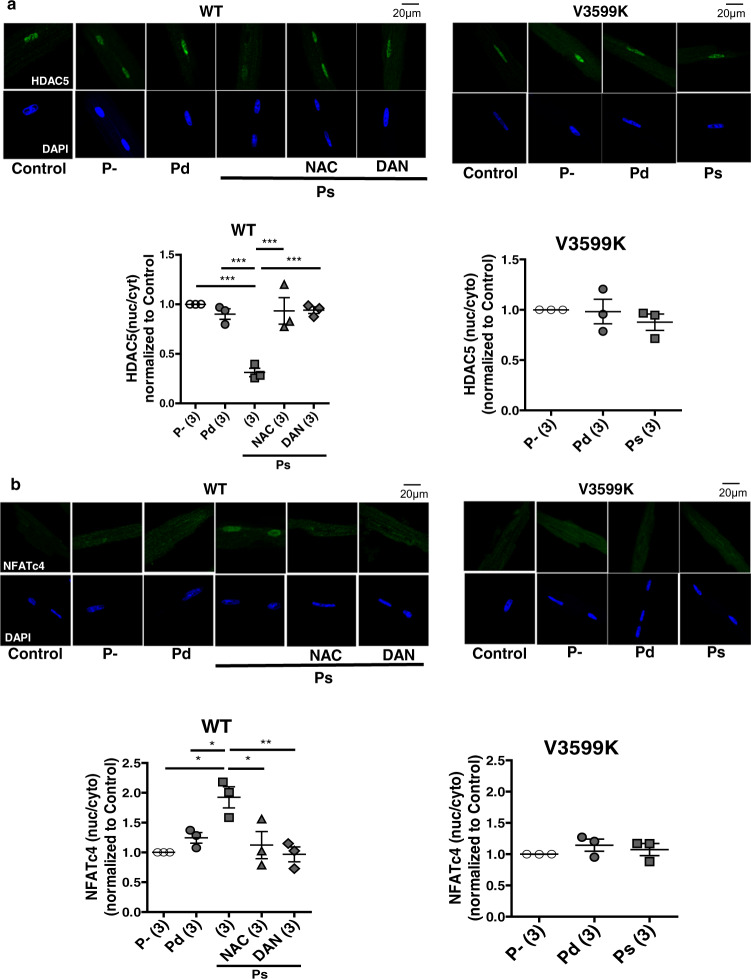
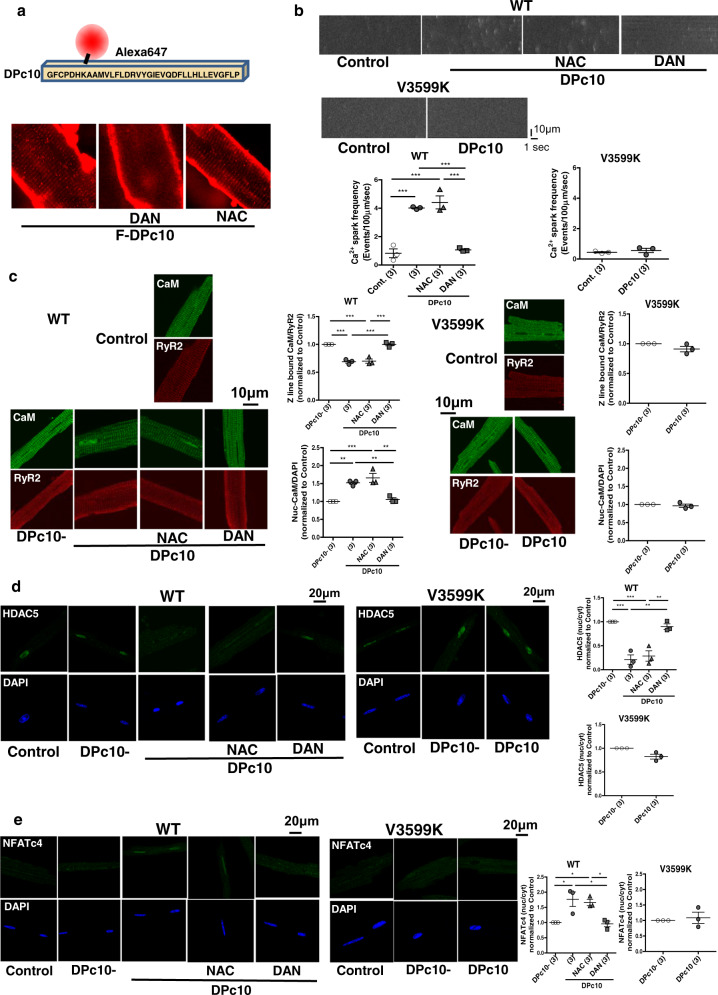
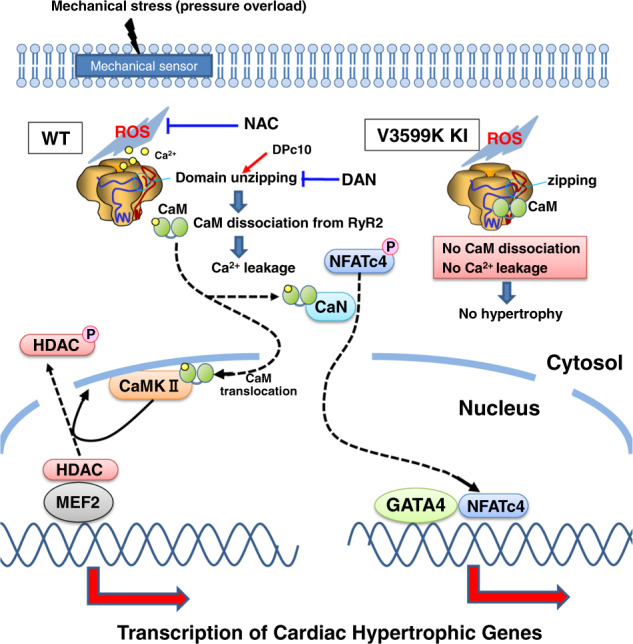
Cardiac hypertrophy is a well-known major risk factor for poor prognosis in patients with cardiovascular diseases. Dysregulation of intracellular Ca2+ is involved in the pathogenesis of cardiac hypertrophy. However, the precise mechanism underlying cardiac hypertrophy remains elusive. Here, we investigate whether pressure-overload induced hypertrophy can be induced by destabilization of cardiac ryanodine receptor (RyR2) through calmodulin (CaM) dissociation and subsequent Ca2+ leakage, and whether it can be genetically rescued by enhancing the binding affinity of CaM to RyR2. In the very initial phase of pressure-overload induced cardiac hypertrophy, when cardiac contractile function is preserved, reactive oxygen species (ROS)-mediated RyR2 destabilization already occurs in association with relaxation dysfunction. Further, stabilizing RyR2 by enhancing the binding affinity of CaM to RyR2 completely inhibits hypertrophic signaling and improves survival. Our study uncovers a critical missing link between RyR2 destabilization and cardiac hypertrophy.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Cardiac hypertrophy associated with impaired regulation of cardiac ryanodine receptor by calmodulin and S100A1.Am J Physiol Heart Circ Physiol. 2013 Jul 1;305(1):H86-94. doi: 10.1152/ajpheart.00144.2013. Epub 2013 May 10. Am J Physiol Heart Circ Physiol. 2013. PMID: 23666671 Free PMC article.
-
Nuclear translocation of calmodulin in pathological cardiac hypertrophy originates from ryanodine receptor bound calmodulin.J Mol Cell Cardiol. 2018 Dec;125:87-97. doi: 10.1016/j.yjmcc.2018.10.011. Epub 2018 Oct 22. J Mol Cell Cardiol. 2018. PMID: 30359562 Free PMC article.
-
The Arrhythmogenic Calmodulin p.Phe142Leu Mutation Impairs C-domain Ca2+ Binding but Not Calmodulin-dependent Inhibition of the Cardiac Ryanodine Receptor.J Biol Chem. 2017 Jan 27;292(4):1385-1395. doi: 10.1074/jbc.M116.766253. Epub 2016 Dec 7. J Biol Chem. 2017. PMID: 27927985 Free PMC article.
-
Molecular regulation of cardiac ryanodine receptor ion channel.Cell Calcium. 2004 Jun;35(6):621-8. doi: 10.1016/j.ceca.2004.01.015. Cell Calcium. 2004. PMID: 15110152 Review.
-
The emerging role of calmodulin regulation of RyR2 in controlling heart rhythm, the progression of heart failure and the antiarrhythmic action of dantrolene.Clin Exp Pharmacol Physiol. 2017 Jan;44(1):135-142. doi: 10.1111/1440-1681.12669. Clin Exp Pharmacol Physiol. 2017. PMID: 27626620 Review.
Cited by
-
NFAT3-FasL axis synchronously regulates apoptosis and necroptosis in murine cochlear outer hair cells after noise trauma.Front Mol Neurosci. 2024 Jul 15;17:1422646. doi: 10.3389/fnmol.2024.1422646. eCollection 2024. Front Mol Neurosci. 2024. PMID: 39077755 Free PMC article.
-
GRP78 Antibodies Are Associated With Blood-Brain Barrier Breakdown in Anti-Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disorder.Neurol Neuroimmunol Neuroinflamm. 2021 Nov 1;9(1):e1038. doi: 10.1212/NXI.0000000000001038. Print 2022 Jan. Neurol Neuroimmunol Neuroinflamm. 2021. PMID: 34725263 Free PMC article.
-
Herpud1 suppress angiotensin II induced hypertrophy in cardiomyocytes.Biochem Biophys Rep. 2022 Mar 16;30:101248. doi: 10.1016/j.bbrep.2022.101248. eCollection 2022 Jul. Biochem Biophys Rep. 2022. PMID: 35313646 Free PMC article.
-
The glycoprotein GPNMB protects against oxidative stress through enhanced PI3K/AKT signaling in epidermal keratinocytes.J Biol Chem. 2025 Mar;301(3):108299. doi: 10.1016/j.jbc.2025.108299. Epub 2025 Feb 11. J Biol Chem. 2025. PMID: 39947468 Free PMC article.
-
Targeting inflammatory macrophages with hyaluronan tetrasaccharide: effects on fibroblast collagen degradation and synthesis.Front Immunol. 2025 Jun 5;16:1592751. doi: 10.3389/fimmu.2025.1592751. eCollection 2025. Front Immunol. 2025. PMID: 40539048 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
