

Single-cell network biology for resolving cellular heterogeneity in human diseases
- PMID: 33244151
- PMCID: PMC8080824
- DOI: 10.1038/s12276-020-00528-0
Single-cell network biology for resolving cellular heterogeneity in human diseases
Abstract
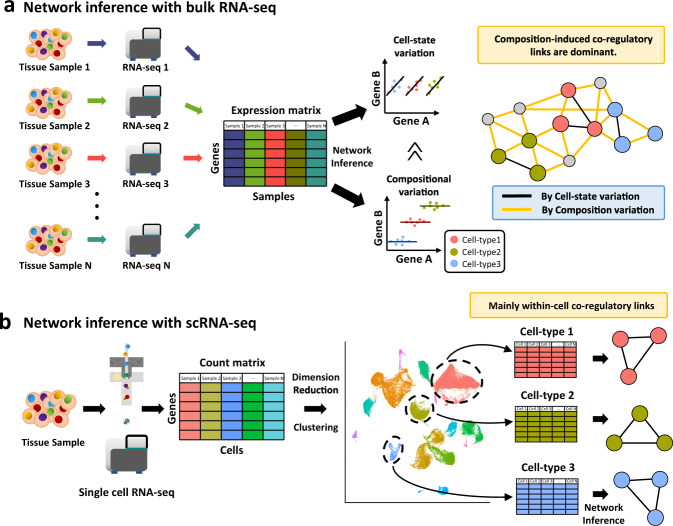
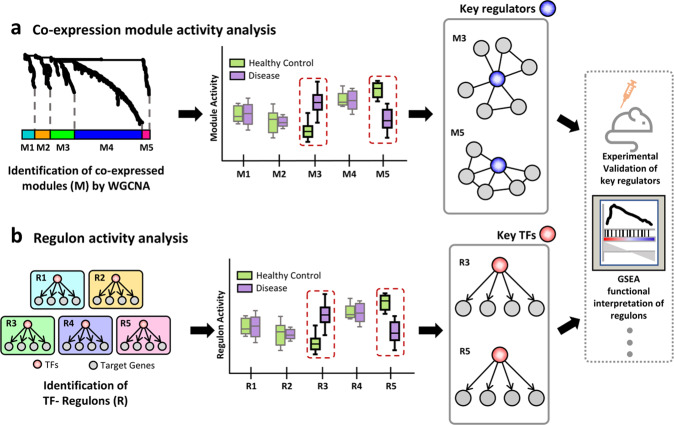
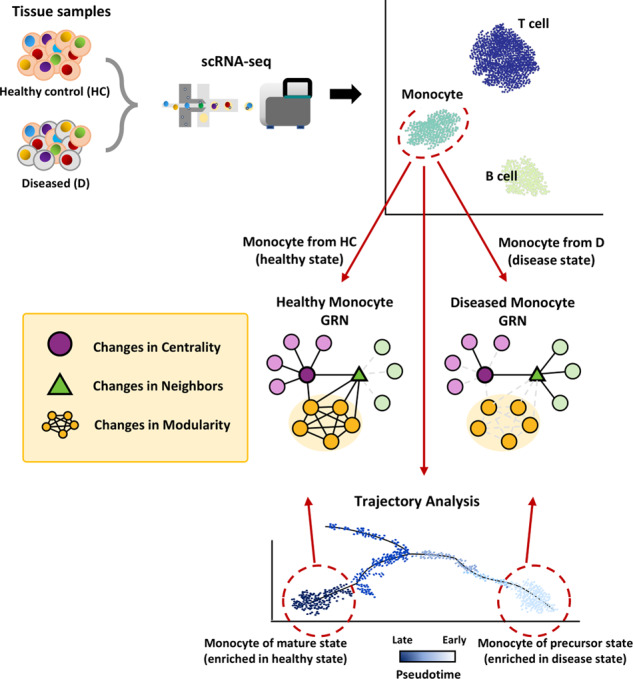
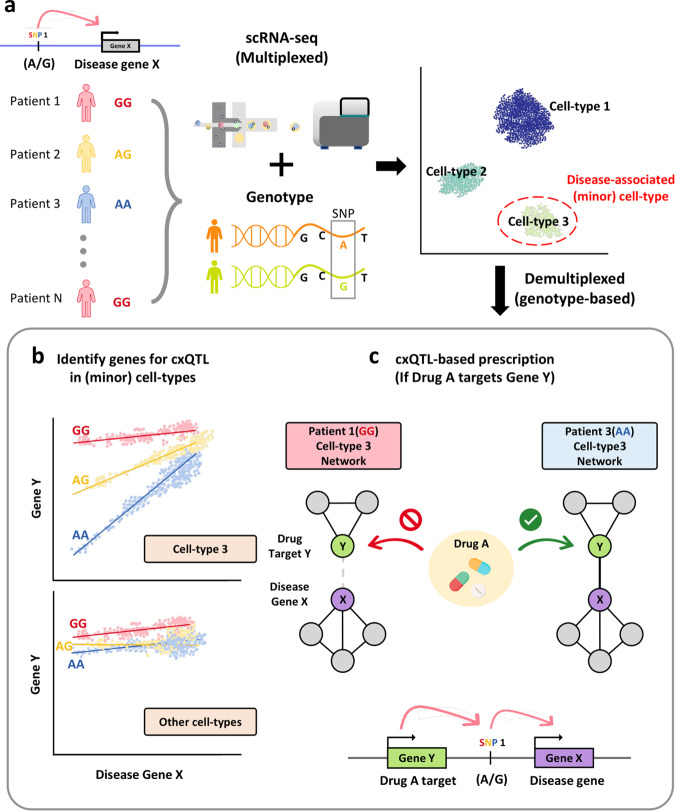
Understanding cellular heterogeneity is the holy grail of biology and medicine. Cells harboring identical genomes show a wide variety of behaviors in multicellular organisms. Genetic circuits underlying cell-type identities will facilitate the understanding of the regulatory programs for differentiation and maintenance of distinct cellular states. Such a cell-type-specific gene network can be inferred from coregulatory patterns across individual cells. Conventional methods of transcriptome profiling using tissue samples provide only average signals of diverse cell types. Therefore, reconstructing gene regulatory networks for a particular cell type is not feasible with tissue-based transcriptome data. Recently, single-cell omics technology has emerged and enabled the capture of the transcriptomic landscape of every individual cell. Although single-cell gene expression studies have already opened up new avenues, network biology using single-cell transcriptome data will further accelerate our understanding of cellular heterogeneity. In this review, we provide an overview of single-cell network biology and summarize recent progress in method development for network inference from single-cell RNA sequencing (scRNA-seq) data. Then, we describe how cell-type-specific gene networks can be utilized to study regulatory programs specific to disease-associated cell types and cellular states. Moreover, with scRNA data, modeling personal or patient-specific gene networks is feasible. Therefore, we also introduce potential applications of single-cell network biology for precision medicine. We envision a rapid paradigm shift toward single-cell network analysis for systems biology in the near future.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Bianconi E, et al. An estimation of the number of cells in the human body. Ann. Hum. Biol. 2013;40:463–471. - PubMed
-
- Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat. Rev. Immunol. 2018;18:35–45. - PubMed
-
- Kolodziejczyk AA, Kim JK, Svensson V, Marioni JC, Teichmann SA. The technology and biology of single-cell RNA sequencing. Mol. Cell. 2015;58:610–620. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
