

The epigenetic basis of cellular heterogeneity
- PMID: 33244170
- PMCID: PMC10880028
- DOI: 10.1038/s41576-020-00300-0
The epigenetic basis of cellular heterogeneity
Abstract
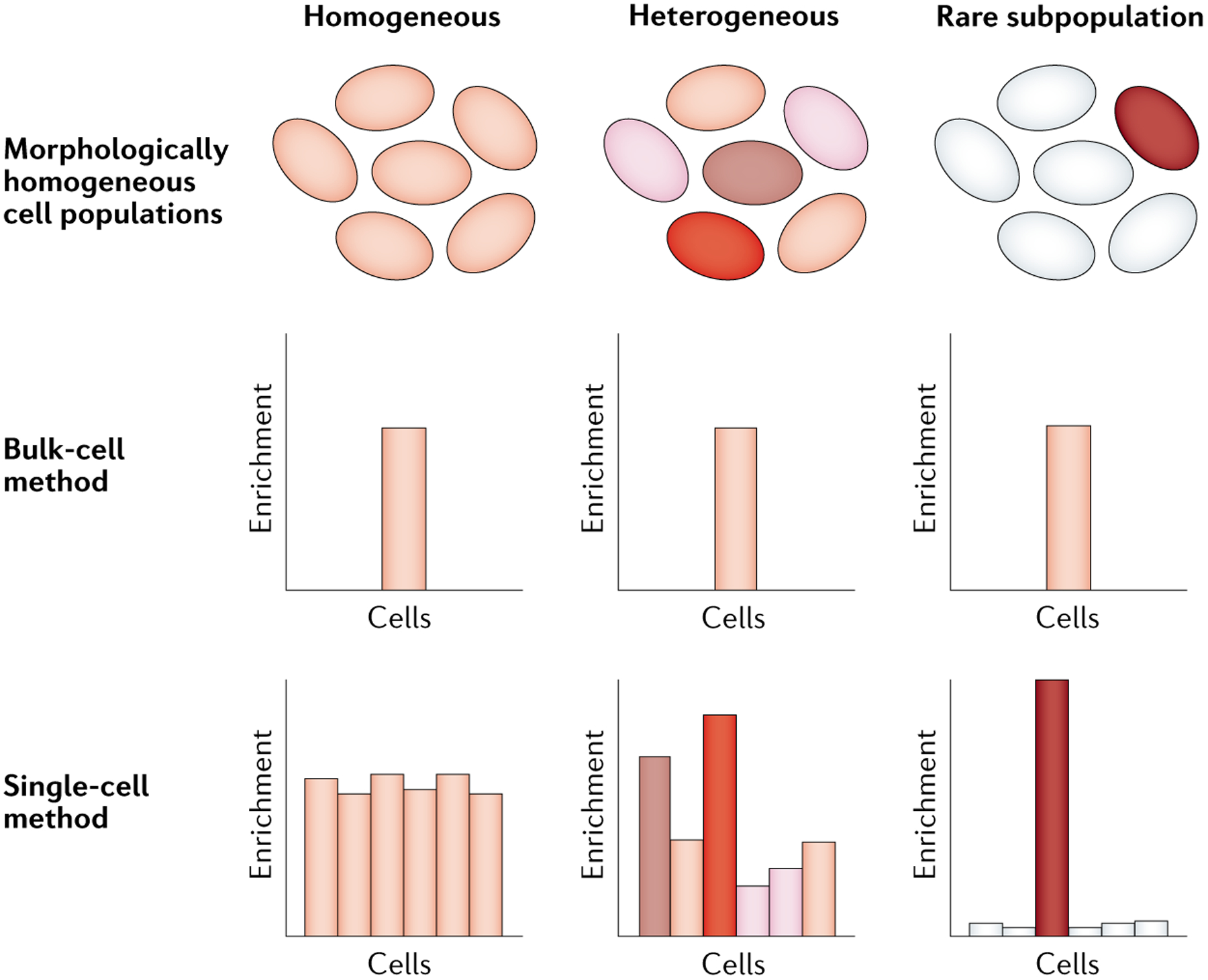
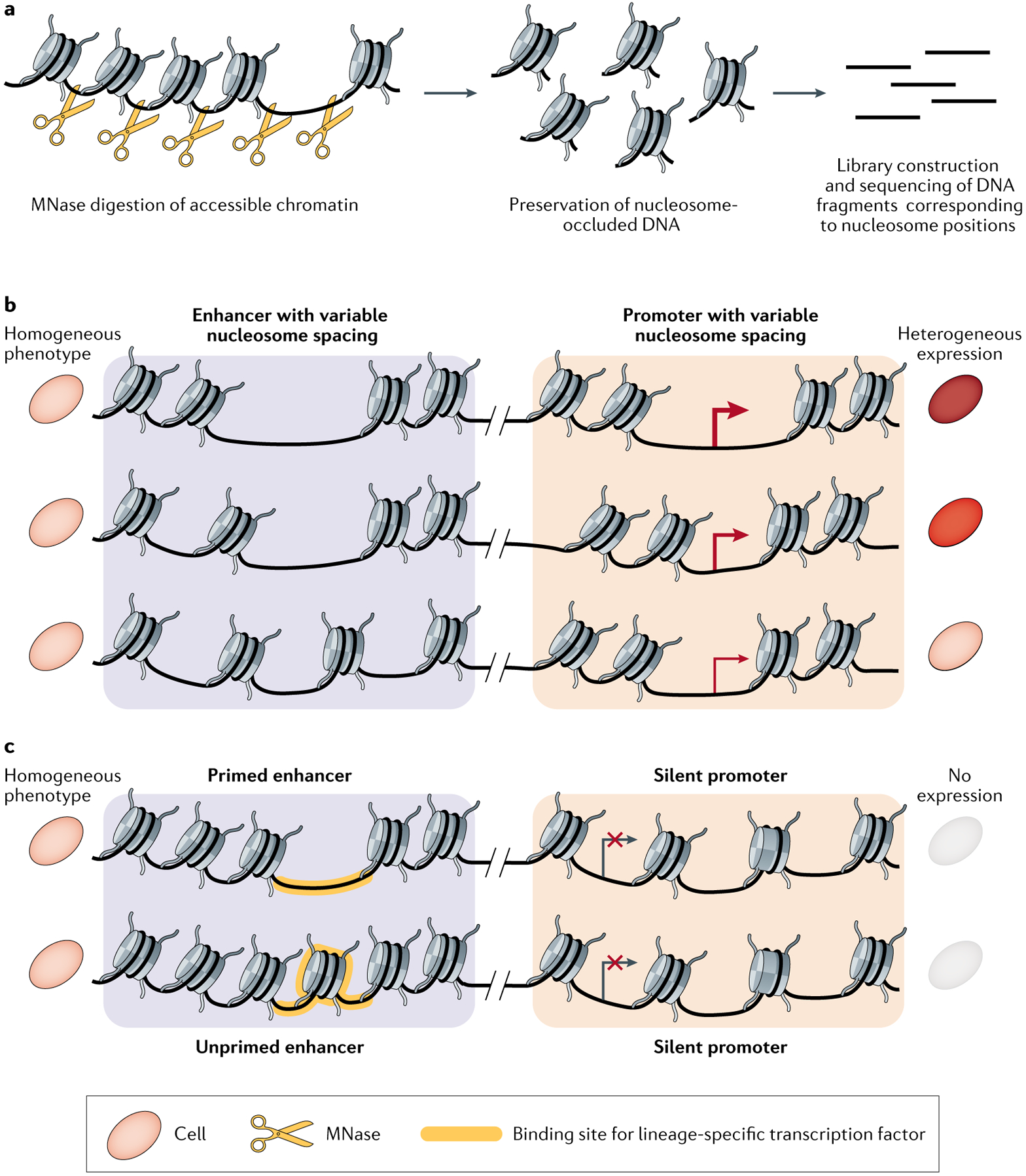
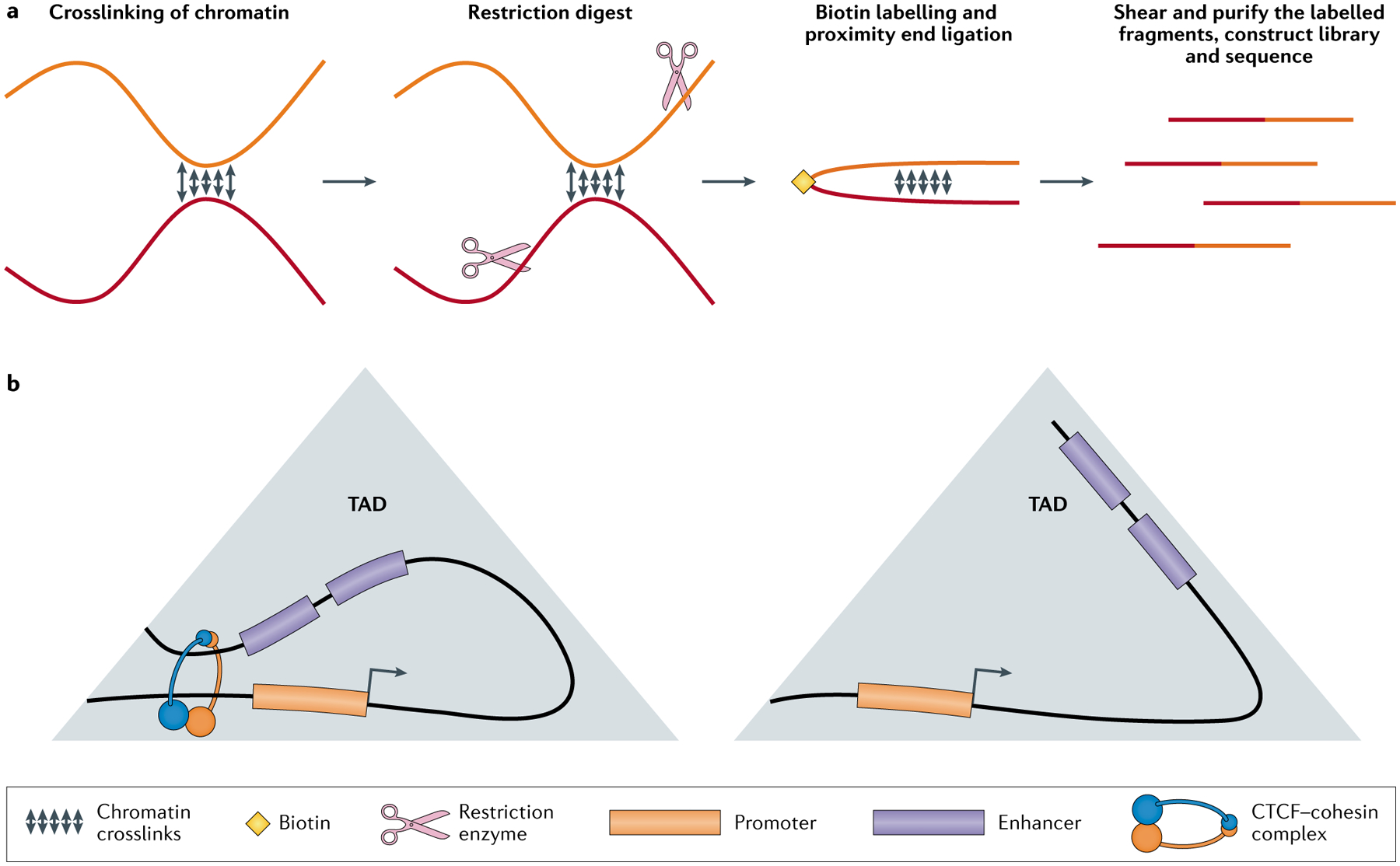
Single-cell sequencing-based methods for profiling gene transcript levels have revealed substantial heterogeneity in expression levels among morphologically indistinguishable cells. This variability has important functional implications for tissue biology and disease states such as cancer. Mapping of epigenomic information such as chromatin accessibility, nucleosome positioning, histone tail modifications and enhancer-promoter interactions in both bulk-cell and single-cell samples has shown that these characteristics of chromatin state contribute to expression or repression of associated genes. Advances in single-cell epigenomic profiling methods are enabling high-resolution mapping of chromatin states in individual cells. Recent studies using these techniques provide evidence that variations in different aspects of chromatin organization collectively define gene expression heterogeneity among otherwise highly similar cells.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures


References
-
- Toyooka Y, Shimosato D, Murakami K, Takahashi K & Niwa H Identification and characterization of subpopulations in undifferentiated ES cell culture. Development 135, 909–918 (2008). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
