

Astrocyte-derived clusterin suppresses amyloid formation in vivo
- PMID: 33246484
- PMCID: PMC7694353
- DOI: 10.1186/s13024-020-00416-1
Astrocyte-derived clusterin suppresses amyloid formation in vivo
Abstract
Background: Accumulation of amyloid-β (Aβ) peptide in the brain is a pathological hallmark of Alzheimer's disease (AD). The clusterin (CLU) gene confers a risk for AD and CLU is highly upregulated in AD patients, with the common non-coding, protective CLU variants associated with increased expression. Although there is strong evidence implicating CLU in amyloid metabolism, the exact mechanism underlying the CLU involvement in AD is not fully understood or whether physiologic alterations of CLU levels in the brain would be protective.
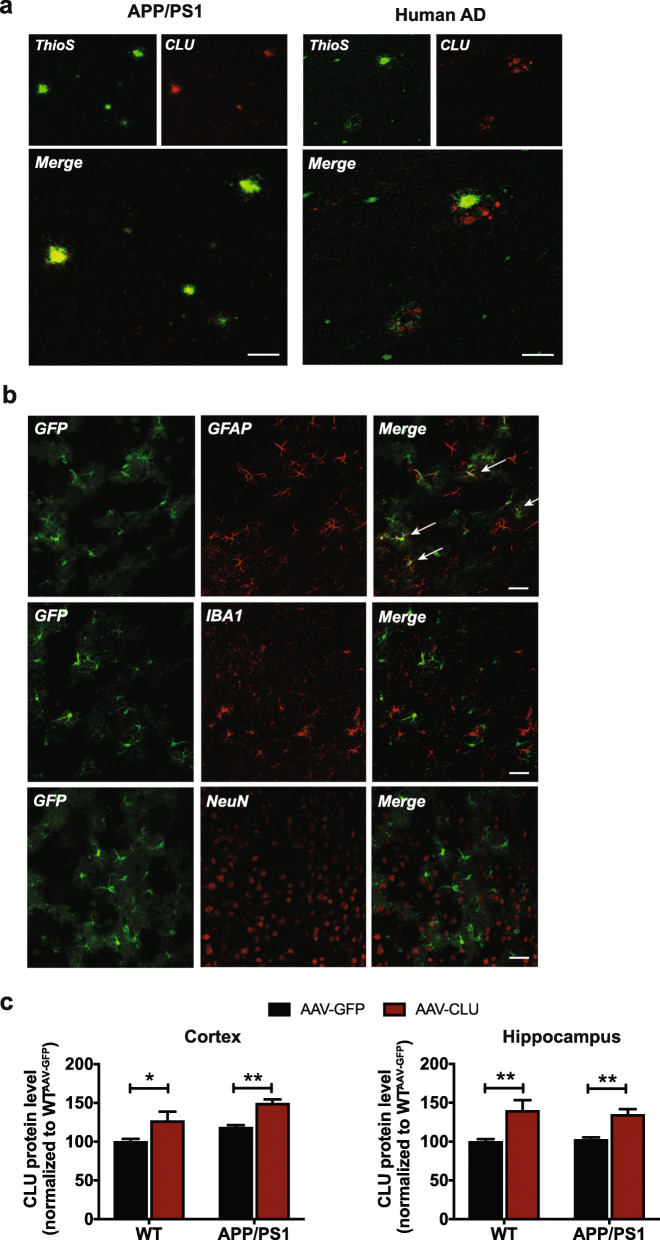
Results: We used a gene delivery approach to overexpress CLU in astrocytes, the major source of CLU expression in the brain. We found that CLU overexpression resulted in a significant reduction of total and fibrillar amyloid in both cortex and hippocampus in the APP/PS1 mouse model of AD amyloidosis. CLU overexpression also ameliorated amyloid-associated neurotoxicity and gliosis. To complement these overexpression studies, we also analyzed the effects of haploinsufficiency of Clu using heterozygous (Clu+/-) mice and control littermates in the APP/PS1 model. CLU reduction led to a substantial increase in the amyloid plaque load in both cortex and hippocampus in APP/PS1; Clu+/- mice compared to wild-type (APP/PS1; Clu+/+) littermate controls, with a concomitant increase in neuritic dystrophy and gliosis.
Conclusions: Thus, both physiologic ~ 30% overexpression or ~ 50% reduction in CLU have substantial impacts on amyloid load and associated pathologies. Our results demonstrate that CLU plays a major role in Aβ accumulation in the brain and suggest that efforts aimed at CLU upregulation via pharmacological or gene delivery approaches offer a promising therapeutic strategy to regulate amyloid pathology.
Keywords: Adeno-associated viral vectors; Alzheimer’s disease; Amyloid plaques; Aβ; Clusterin; Haploinsufficiency.
Conflict of interest statement
The authors declare that no competing interests exist.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
