

In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo
- PMID: 33247092
- PMCID: PMC7695830
- DOI: 10.1038/s41467-020-19486-2
In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo
Abstract
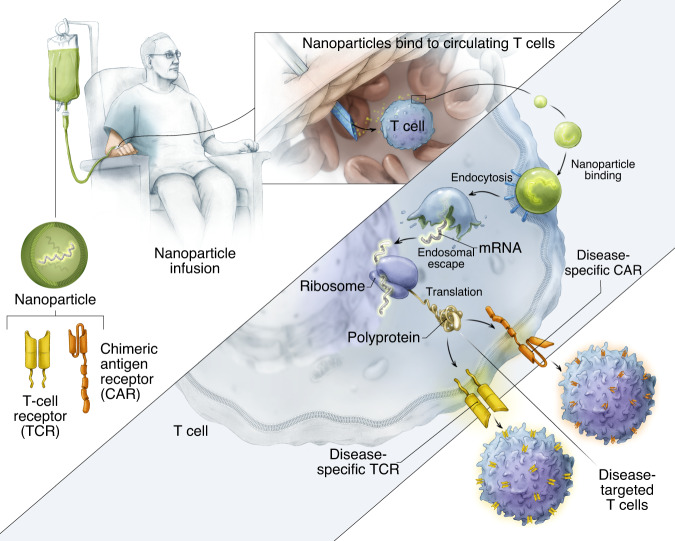
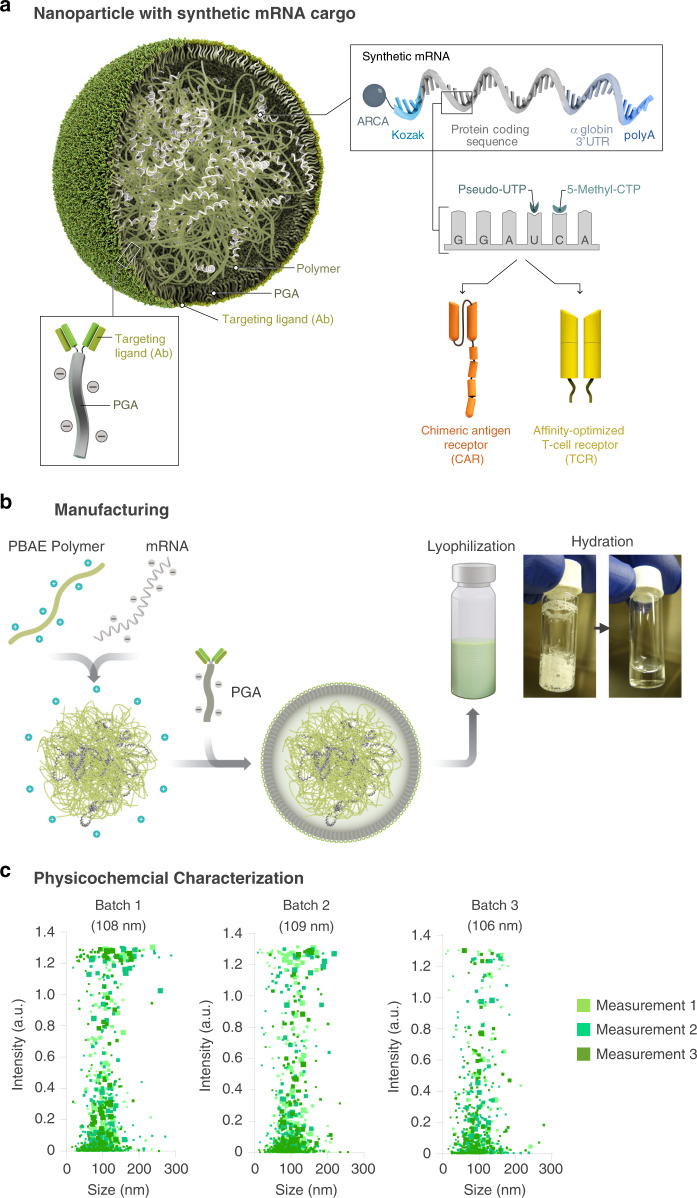
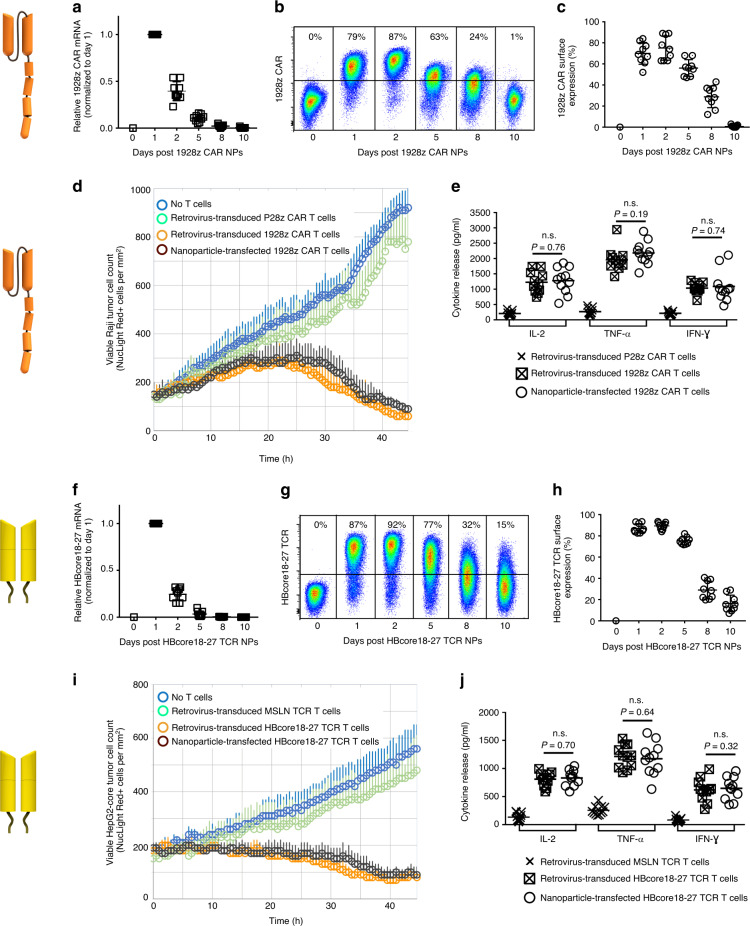
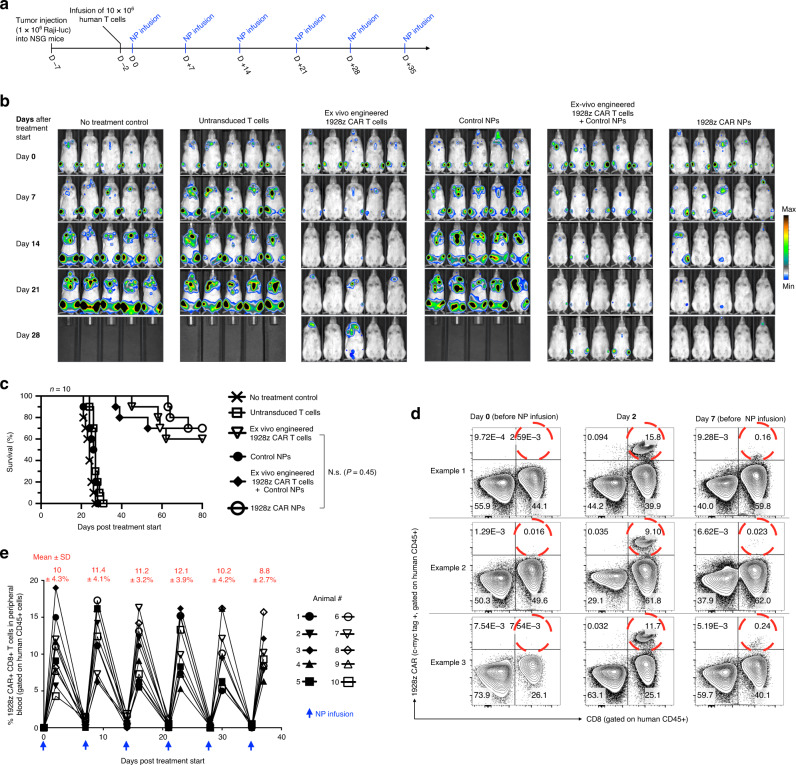
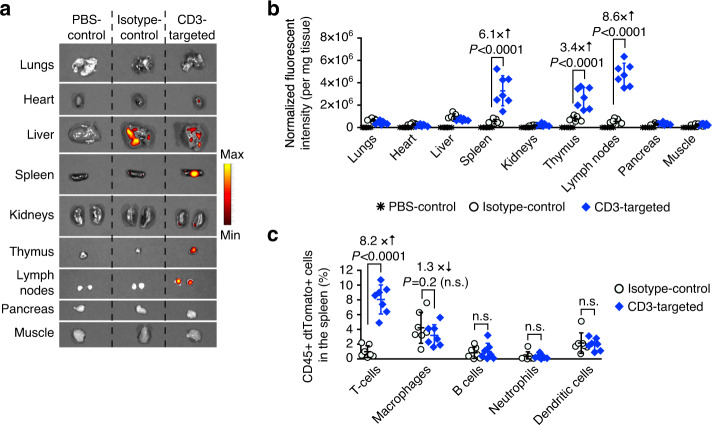
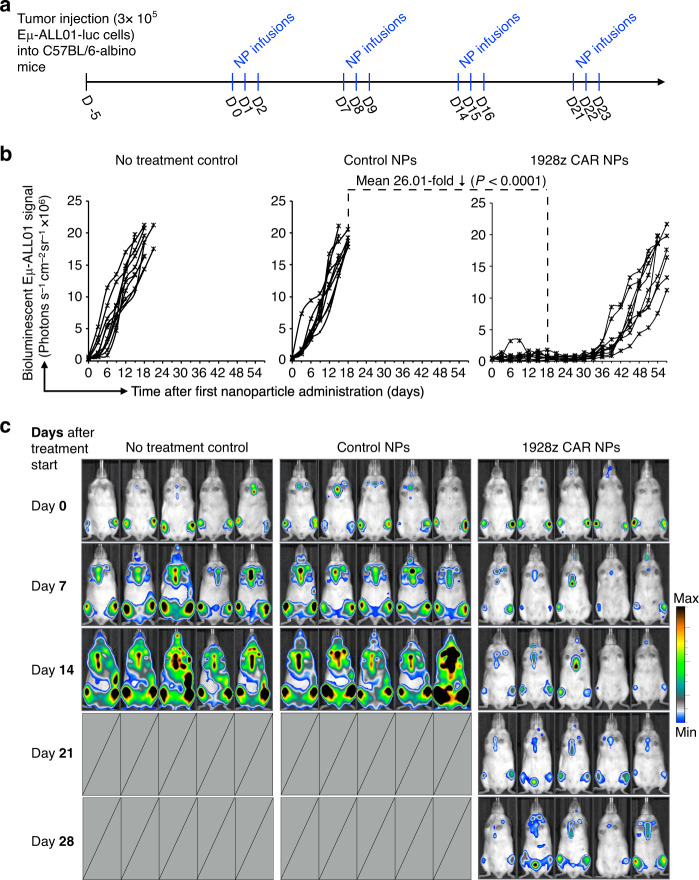
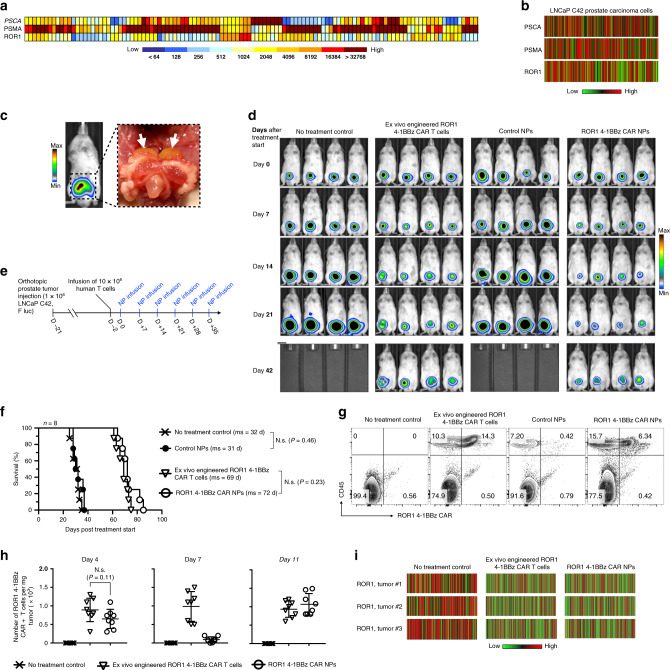
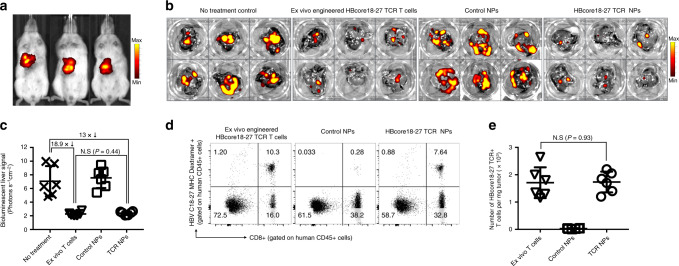
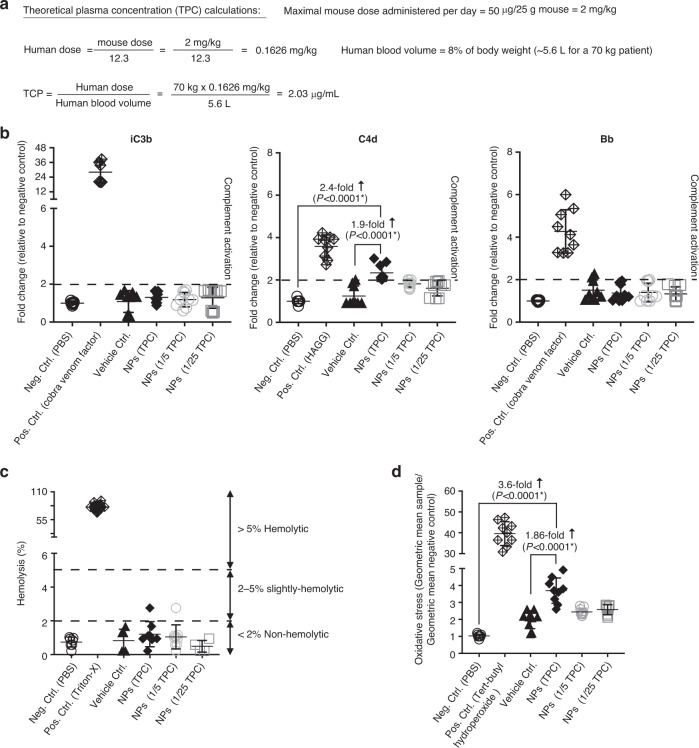
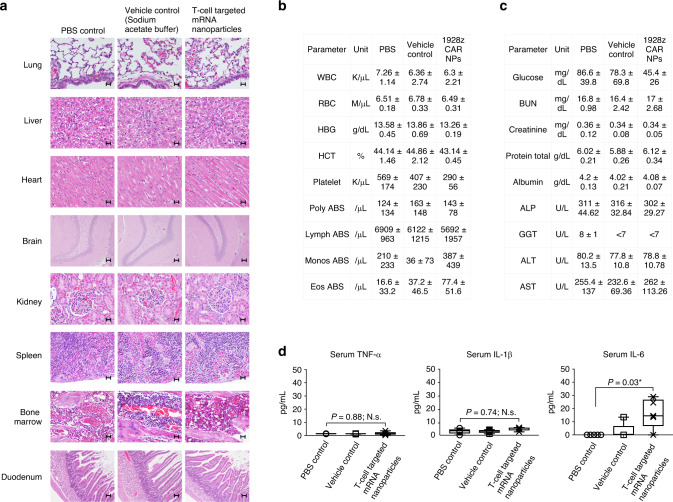
Engineering chimeric antigen receptors (CAR) or T cell receptors (TCR) helps create disease-specific T cells for targeted therapy, but the cost and rigor associated with manufacturing engineered T cells ex vivo can be prohibitive, so programing T cells in vivo may be a viable alternative. Here we report an injectable nanocarrier that delivers in vitro-transcribed (IVT) CAR or TCR mRNA for transiently reprograming of circulating T cells to recognize disease-relevant antigens. In mouse models of human leukemia, prostate cancer and hepatitis B-induced hepatocellular carcinoma, repeated infusions of these polymer nanocarriers induce sufficient host T cells expressing tumor-specific CARs or virus-specific TCRs to cause disease regression at levels similar to bolus infusions of ex vivo engineered lymphocytes. Given their ease of manufacturing, distribution and administration, these nanocarriers, and the associated platforms, could become a therapeutic for a wide range of diseases.
Conflict of interest statement
M.T.S. is a consultant of Tidal Therapeutics and holds stocks in the company. The remaining authors declare no competing interests.
Figures
References
-
- Rockoff JD. The million-dollar cancer treatment: who will pay? Wall Str. J. 2018;July 10:2018.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
