

An inducible CRISPR interference library for genetic interrogation of Saccharomyces cerevisiae biology
- PMID: 33247197
- PMCID: PMC7695836
- DOI: 10.1038/s42003-020-01452-9
An inducible CRISPR interference library for genetic interrogation of Saccharomyces cerevisiae biology
Abstract
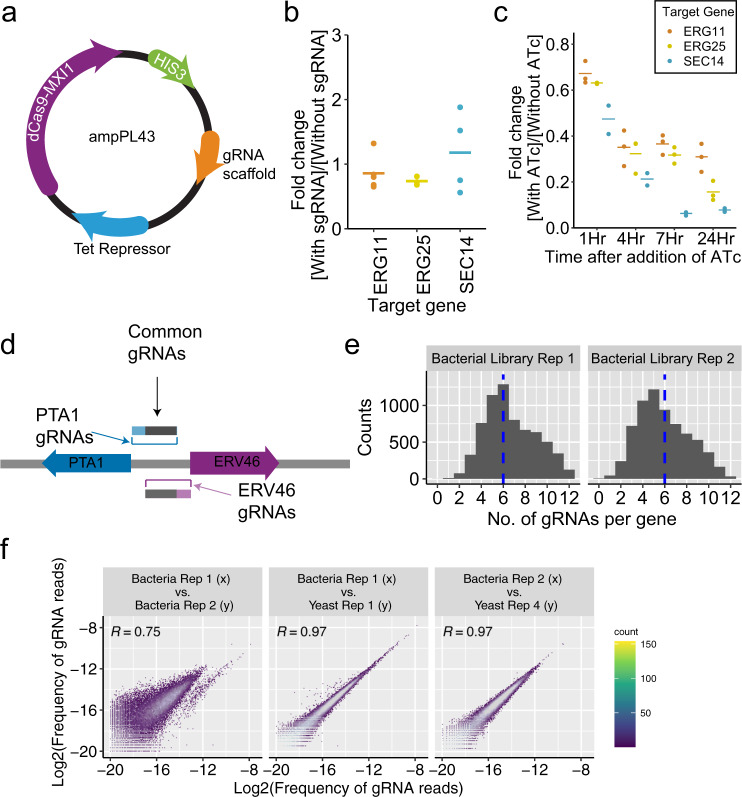
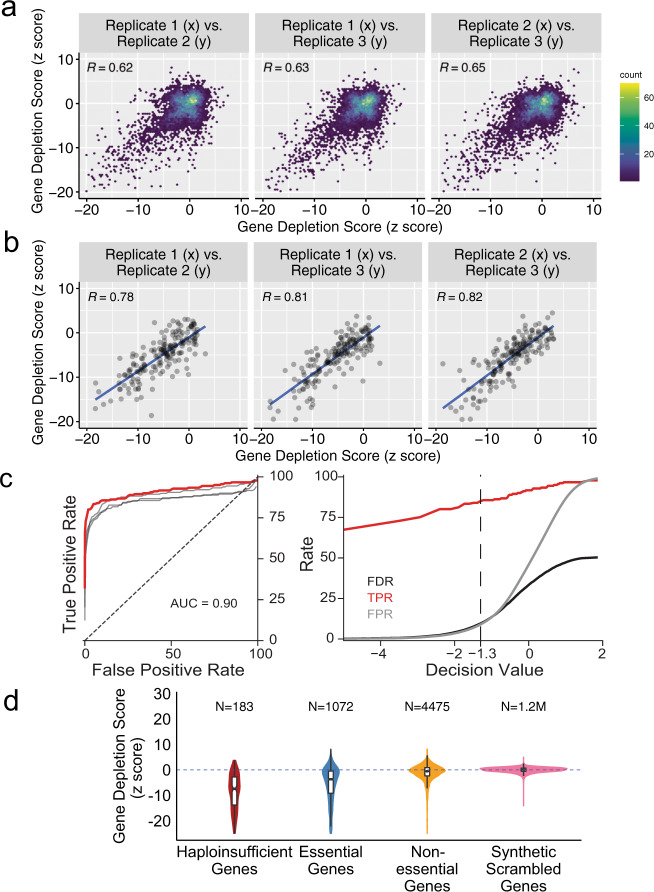
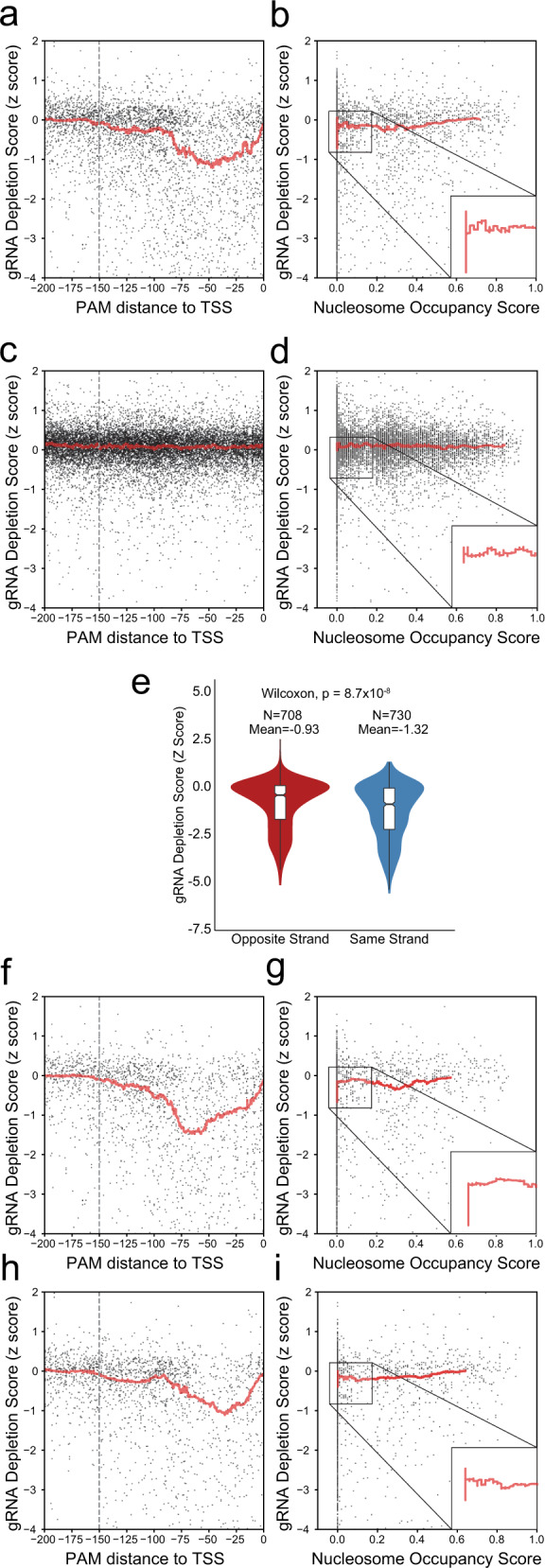
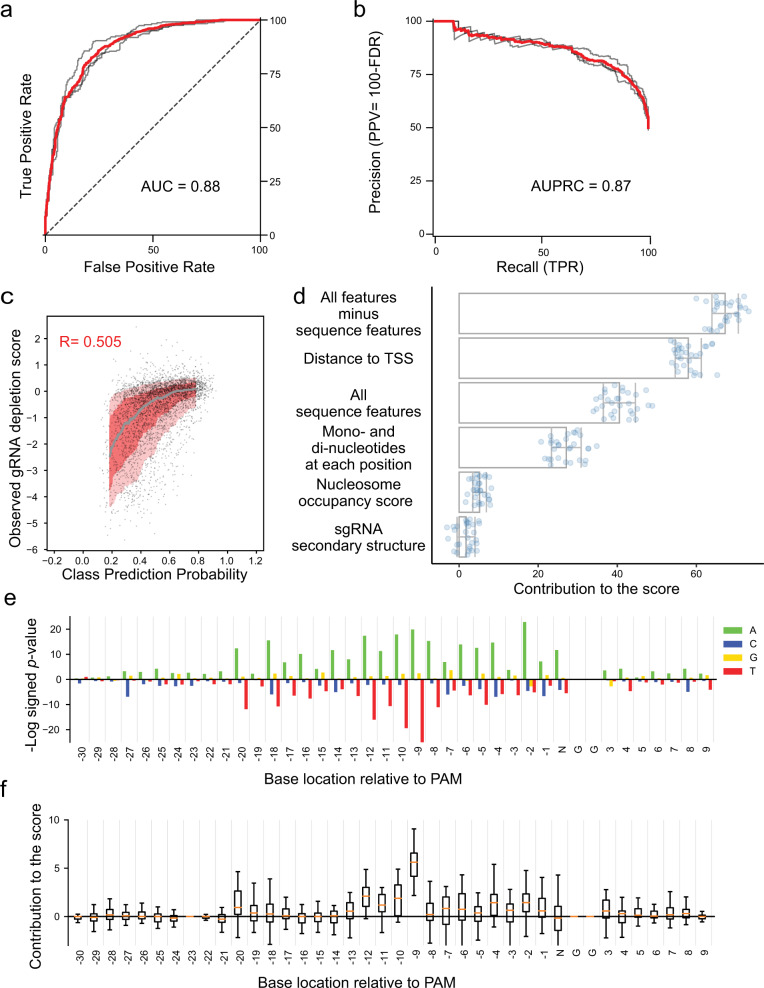
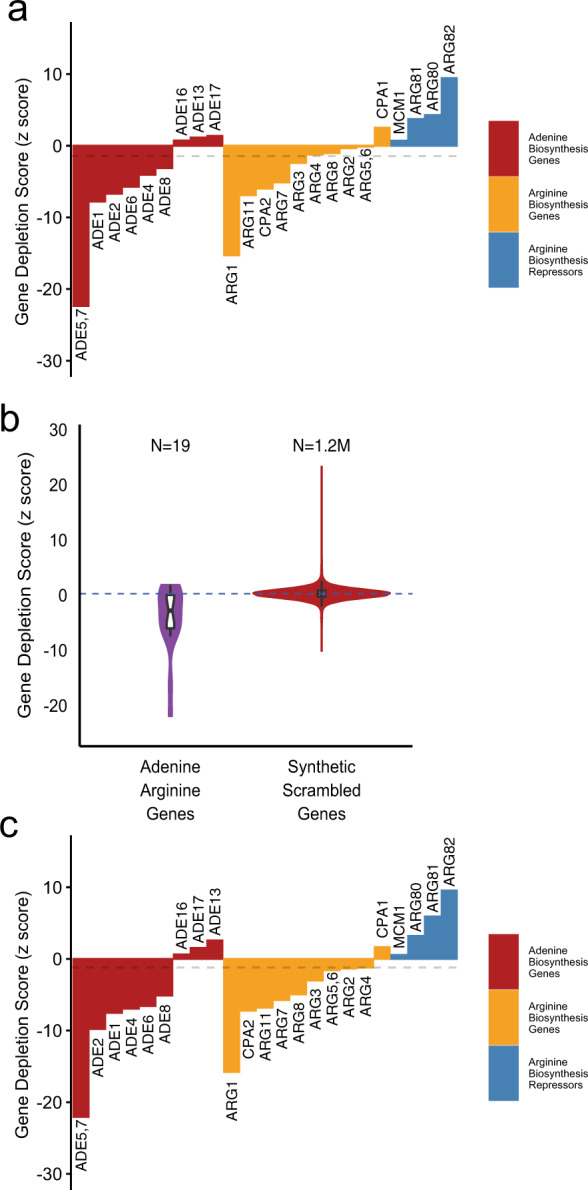
Genome-scale CRISPR interference (CRISPRi) is widely utilized to study cellular processes in a variety of organisms. Despite the dominance of Saccharomyces cerevisiae as a model eukaryote, an inducible genome-wide CRISPRi library in yeast has not yet been presented. Here, we present a genome-wide, inducible CRISPRi library, based on spacer design rules optimized for S. cerevisiae. We have validated this library for genome-wide interrogation of gene function across a variety of applications, including accurate discovery of haploinsufficient genes and identification of enzymatic and regulatory genes involved in adenine and arginine biosynthesis. The comprehensive nature of the library also revealed refined spacer design parameters for transcriptional repression, including location, nucleosome occupancy and nucleotide features. CRISPRi screens using this library can identify genes and pathways with high precision and a low false discovery rate across a variety of experimental conditions, enabling rapid and reliable assessment of genetic function and interactions in S. cerevisiae.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
A genome-scale CRISPR interference guide library enables comprehensive phenotypic profiling in yeast.BMC Genomics. 2021 Mar 23;22(1):205. doi: 10.1186/s12864-021-07518-0. BMC Genomics. 2021. PMID: 33757429 Free PMC article.
-
Quantitative CRISPR interference screens in yeast identify chemical-genetic interactions and new rules for guide RNA design.Genome Biol. 2016 Mar 8;17:45. doi: 10.1186/s13059-016-0900-9. Genome Biol. 2016. PMID: 26956608 Free PMC article.
-
A genome-scale yeast library with inducible expression of individual genes.Mol Syst Biol. 2021 Jun;17(6):e10207. doi: 10.15252/msb.202110207. Mol Syst Biol. 2021. PMID: 34096681 Free PMC article.
-
Yeast genetic interaction screens in the age of CRISPR/Cas.Curr Genet. 2019 Apr;65(2):307-327. doi: 10.1007/s00294-018-0887-8. Epub 2018 Sep 25. Curr Genet. 2019. PMID: 30255296 Free PMC article. Review.
-
CRISPR/Cas system for yeast genome engineering: advances and applications.FEMS Yeast Res. 2017 Aug 1;17(5):fox030. doi: 10.1093/femsyr/fox030. FEMS Yeast Res. 2017. PMID: 28505256 Free PMC article. Review.
Cited by
-
Identification of acetic acid sensitive strains through biosensor-based screening of a Saccharomyces cerevisiae CRISPRi library.Microb Cell Fact. 2022 Oct 15;21(1):214. doi: 10.1186/s12934-022-01938-7. Microb Cell Fact. 2022. PMID: 36243715 Free PMC article.
-
Recent Advances in Directed Yeast Genome Evolution.J Fungi (Basel). 2022 Jun 15;8(6):635. doi: 10.3390/jof8060635. J Fungi (Basel). 2022. PMID: 35736118 Free PMC article. Review.
-
CRISPRi screens reveal genes modulating yeast growth in lignocellulose hydrolysate.Biotechnol Biofuels. 2021 Feb 10;14(1):41. doi: 10.1186/s13068-021-01880-7. Biotechnol Biofuels. 2021. PMID: 33568224 Free PMC article.
-
Transcriptional Regulation Technology for Gene Perturbation in Fission Yeast.Biomolecules. 2023 Apr 21;13(4):716. doi: 10.3390/biom13040716. Biomolecules. 2023. PMID: 37189462 Free PMC article. Review.
-
Yca1 metacaspase: diverse functions determine how yeast live and let die.FEMS Yeast Res. 2023 Jan 4;23:foad022. doi: 10.1093/femsyr/foad022. FEMS Yeast Res. 2023. PMID: 37002543 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
