

Docking-based identification of small-molecule binding sites at protein-protein interfaces
- PMID: 33250973
- PMCID: PMC7679229
- DOI: 10.1016/j.csbj.2020.11.029
Docking-based identification of small-molecule binding sites at protein-protein interfaces
Abstract
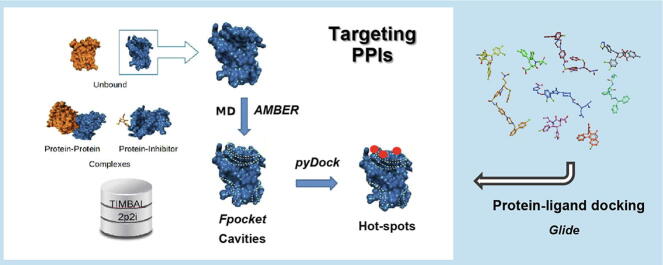
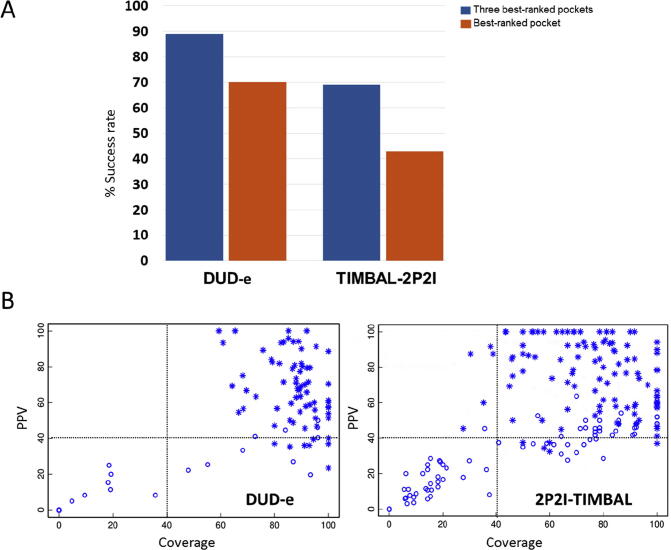
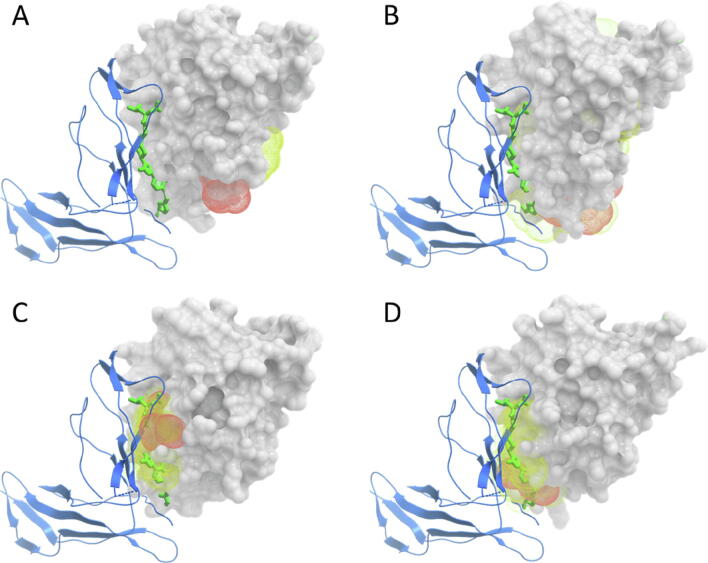
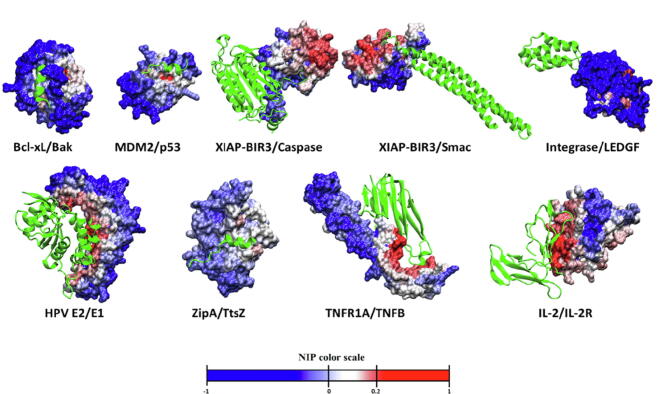
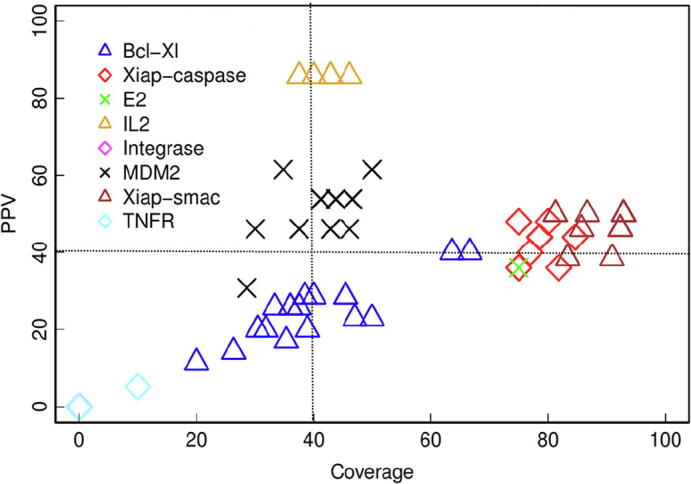
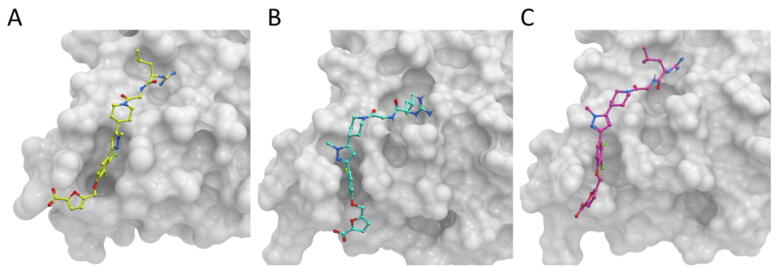
Protein-protein interactions play an essential role in many biological processes, and their perturbation is a major cause of disease. The use of small molecules to modulate them is attracting increased attention, but protein interfaces generally do not have clear cavities for binding small compounds. A proposed strategy is to target interface hot-spot residues, but their identification through computational approaches usually require the complex structure, which is not often available. In this context, pyDock energy-based docking and scoring can predict hot-spots on the unbound proteins, thus not requiring the complex structure. Here, we have devised a new strategy to detect protein-protein inhibitor binding sites, based on the integration of molecular dynamics for the generation of transient cavities, and docking-based interface hot-spot prediction for the selection of the suitable cavities. This integrative approach has been validated on a test set formed by protein-protein complexes with known inhibitors for which complete structural data of unbound molecules and complexes is available. The results show that local conformational sampling with short molecular dynamics can generate transient cavities similar to the known inhibitor binding sites, and that docking simulations can identify the best cavities with similar predictive accuracy as when knowing the real interface. In a few cases, these predicted pockets are shown to be suitable for protein-ligand docking. The proposed strategy will be useful for many protein-protein complexes for which there is no available structure, as long as the the unbound proteins do not deviate dramatically from the bound conformations.
Keywords: Cavity detection; Drug discovery; Interface hot-spot residues; Modulation of protein–protein interactions; Molecular dynamics; Protein docking simulations.
© 2020 The Author(s).
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures
Similar articles
-
Hot-spot analysis for drug discovery targeting protein-protein interactions.Expert Opin Drug Discov. 2018 Apr;13(4):327-338. doi: 10.1080/17460441.2018.1430763. Epub 2018 Jan 29. Expert Opin Drug Discov. 2018. PMID: 29376444 Review.
-
Hot spots and transient pockets: predicting the determinants of small-molecule binding to a protein-protein interface.J Chem Inf Model. 2012 Jan 23;52(1):120-33. doi: 10.1021/ci200322s. Epub 2011 Dec 27. J Chem Inf Model. 2012. PMID: 22087639
-
Identification of hot-spot residues in protein-protein interactions by computational docking.BMC Bioinformatics. 2008 Oct 21;9:447. doi: 10.1186/1471-2105-9-447. BMC Bioinformatics. 2008. PMID: 18939967 Free PMC article.
-
Hot spots in protein-protein interfaces: towards drug discovery.Prog Biophys Mol Biol. 2014 Nov-Dec;116(2-3):165-73. doi: 10.1016/j.pbiomolbio.2014.06.003. Epub 2014 Jul 2. Prog Biophys Mol Biol. 2014. PMID: 24997383 Review.
-
Targeting protein-protein interactions with small molecules: challenges and perspectives for computational binding epitope detection and ligand finding.Curr Med Chem. 2006;13(22):2607-25. doi: 10.2174/092986706778201530. Curr Med Chem. 2006. PMID: 17017914 Review.
Cited by
-
Towards design of drugs and delivery systems with the Martini coarse-grained model.QRB Discov. 2022 Oct 12;3:e19. doi: 10.1017/qrd.2022.16. eCollection 2022. QRB Discov. 2022. PMID: 37529288 Free PMC article. Review.
-
Resources and computational strategies to advance small molecule SARS-CoV-2 discovery: Lessons from the pandemic and preparing for future health crises.Comput Struct Biotechnol J. 2021;19:2537-2548. doi: 10.1016/j.csbj.2021.04.059. Epub 2021 Apr 26. Comput Struct Biotechnol J. 2021. PMID: 33936562 Free PMC article. Review.
-
Protein-protein interaction prediction methods: from docking-based to AI-based approaches.Biophys Rev. 2022 Dec 17;14(6):1341-1348. doi: 10.1007/s12551-022-01032-7. eCollection 2022 Dec. Biophys Rev. 2022. PMID: 36570321 Free PMC article. Review.
-
Delineating the conformational landscape and intrinsic properties of the angiotensin II type 2 receptor using a computational study.Comput Struct Biotechnol J. 2022 May 10;20:2268-2279. doi: 10.1016/j.csbj.2022.05.012. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 35615027 Free PMC article.
-
Mapping the binding sites of challenging drug targets.Curr Opin Struct Biol. 2022 Aug;75:102396. doi: 10.1016/j.sbi.2022.102396. Epub 2022 May 27. Curr Opin Struct Biol. 2022. PMID: 35636004 Free PMC article. Review.
References
-
- Rosell M., Fernández-Recio J. Hot-spot analysis for drug discovery targeting protein-protein interactions. Expert Opin Drug Discov. 2018;13(4):327–338. - PubMed
LinkOut - more resources
Full Text Sources