

Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic
- PMID: 33251389
- PMCID: PMC7680079
- DOI: 10.1186/s13765-020-00564-4
Screening marine algae metabolites as high-affinity inhibitors of SARS-CoV-2 main protease (3CLpro): an in silico analysis to identify novel drug candidates to combat COVID-19 pandemic
Abstract
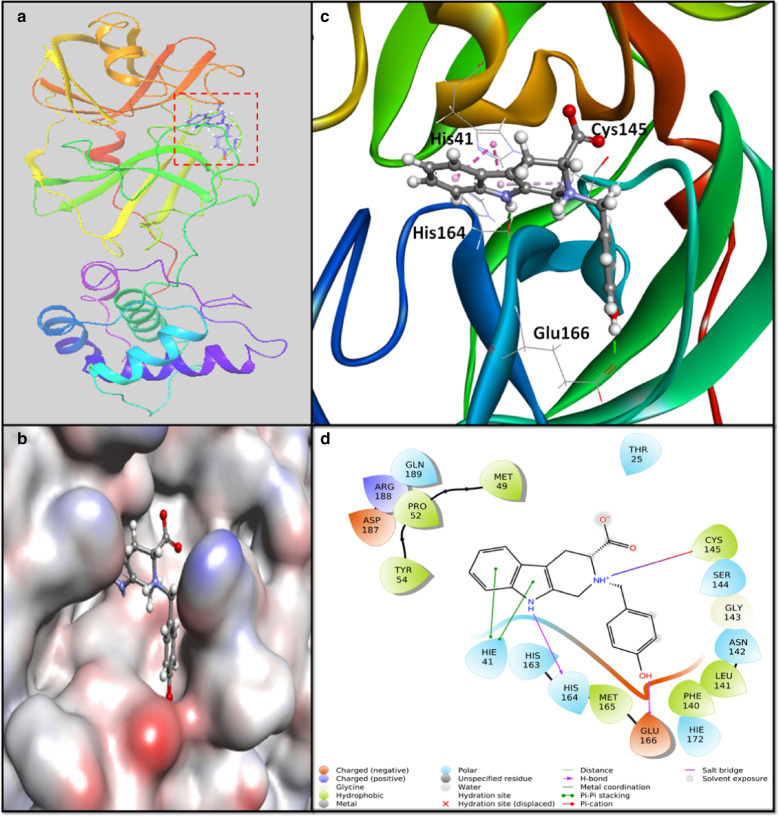
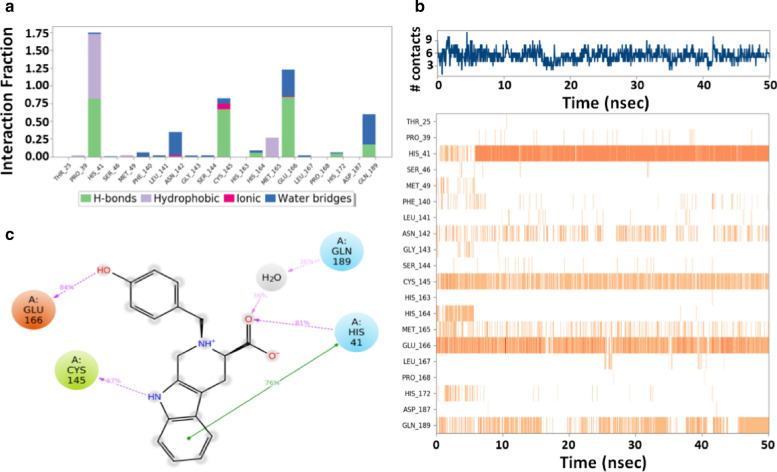
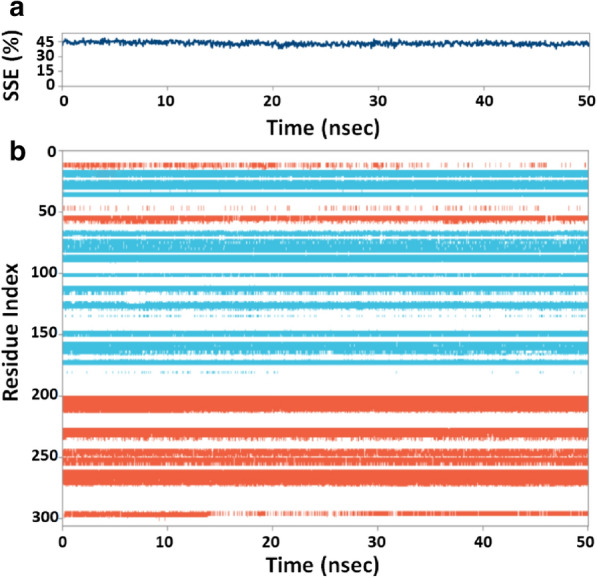
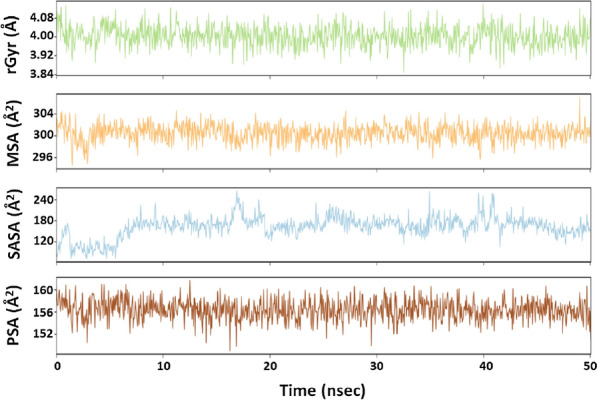
The recent dissemination of SARS-CoV-2 from Wuhan city to all over the world has created a pandemic. COVID-19 has cost many human lives and created an enormous economic burden. Although many drugs/vaccines are in different stages of clinical trials, still none is clinically available. We have screened a marine seaweed database (1110 compounds) against 3CLpro of SARS-CoV-2 using computational approaches. High throughput virtual screening was performed on compounds, and 86 of them with docking score < - 5.000 kcal mol-1 were subjected to standard-precision docking. Based on binding energies (< - 6.000 kcal mol-1), 9 compounds were further shortlisted and subjected to extra-precision docking. Free energy calculation by Prime-MM/GBSA suggested RC002, GA004, and GA006 as the most potent inhibitors of 3CLpro. An analysis of ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties of RC002, GA004, and GA006 indicated that only RC002 (callophysin A, from red alga Callophycus oppositifolius) passed Lipinski's, Veber's, PAINS and Brenk's filters and displayed drug-like and lead-like properties. Analysis of 3CLpro-callophysin A complex revealed the involvement of salt bridge, hydrogen bonds, and hydrophobic interactions. callophysin A interacted with the catalytic residues (His41 and Cys145) of 3CLpro; hence it may act as a mechanism-based competitive inhibitor. Docking energy and docking affinity of callophysin A towards 3CLpro was - 8.776 kcal mol-1 and 2.73 × 106 M-1, respectively. Molecular dynamics simulation confirmed the stability of the 3CLpro-callophysin A complex. The findings of this study may serve as the basis for further validation by in vitro and in vivo studies.
Keywords: Callophysin A; Marine-derived compounds; Molecular docking and simulation; SARS-CoV-2; Seaweeds.
© The Author(s) 2020.
Conflict of interest statement
Competing interestsAll the authors declare that there is no competing interest that are relevant to the content of this article.
Figures

Similar articles
-
Identification of natural compounds (proanthocyanidin and rhapontin) as high-affinity inhibitors of SARS-CoV-2 Mpro and PLpro using computational strategies.Arch Med Sci. 2021 Mar 20;20(2):567-581. doi: 10.5114/aoms/133706. eCollection 2024. Arch Med Sci. 2021. PMID: 38757037 Free PMC article.
-
Natural Compounds as Inhibitors of SARS-CoV-2 Main Protease (3CLpro): A Molecular Docking and Simulation Approach to Combat COVID-19.Curr Pharm Des. 2021;27(33):3577-3589. doi: 10.2174/1381612826999201116195851. Curr Pharm Des. 2021. PMID: 33200697
-
Discovery of putative inhibitors against main drivers of SARS-CoV-2 infection: Insight from quantum mechanical evaluation and molecular modeling.Front Chem. 2022 Oct 11;10:964446. doi: 10.3389/fchem.2022.964446. eCollection 2022. Front Chem. 2022. PMID: 36304744 Free PMC article.
-
A comprehensive review on promising anti-viral therapeutic candidates identified against main protease from SARS-CoV-2 through various computational methods.J Genet Eng Biotechnol. 2020 Nov 3;18(1):69. doi: 10.1186/s43141-020-00085-z. J Genet Eng Biotechnol. 2020. PMID: 33141358 Free PMC article. Review.
-
Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale.RSC Adv. 2020 May 27;10(34):19790-19802. doi: 10.1039/d0ra04199h. eCollection 2020 May 26. RSC Adv. 2020. PMID: 35685913 Free PMC article. Review.
Cited by
-
Concatenation of molecular docking and molecular simulation of BACE-1, γ-secretase targeted ligands: in pursuit of Alzheimer's treatment.Ann Med. 2021 Dec;53(1):2332-2344. doi: 10.1080/07853890.2021.2009124. Ann Med. 2021. PMID: 34889159 Free PMC article.
-
Systematic Search for SARS-CoV-2 Main Protease Inhibitors for Drug Repurposing: Ethacrynic Acid as a Potential Drug.Viruses. 2021 Jan 13;13(1):106. doi: 10.3390/v13010106. Viruses. 2021. PMID: 33451132 Free PMC article.
-
Soyasapogenol-B as a Potential Multitarget Therapeutic Agent for Neurodegenerative Disorders: Molecular Docking and Dynamics Study.Entropy (Basel). 2022 Apr 23;24(5):593. doi: 10.3390/e24050593. Entropy (Basel). 2022. PMID: 35626478 Free PMC article.
-
Unravelling the therapeutic potential of marine drugs as SARS-CoV-2 inhibitors: An insight from essential dynamics and free energy landscape.Comput Biol Med. 2021 Aug;135:104525. doi: 10.1016/j.compbiomed.2021.104525. Epub 2021 May 29. Comput Biol Med. 2021. PMID: 34252682 Free PMC article.
-
C60 fullerene against SARS-CoV-2 coronavirus: an in silico insight.Sci Rep. 2021 Sep 7;11(1):17748. doi: 10.1038/s41598-021-97268-6. Sci Rep. 2021. PMID: 34493768 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Miscellaneous