Enhanced mitophagy in bronchial fibroblasts from severe asthmatic patients
- PMID: 33253229
- PMCID: PMC7704010
- DOI: 10.1371/journal.pone.0242695
Enhanced mitophagy in bronchial fibroblasts from severe asthmatic patients
Abstract
Background: Sub-epithelial fibrosis is a characteristic feature of airway remodeling in asthma which correlates with disease severity. Current asthma medications are ineffective in treating fibrosis. In this study, we aimed to investigate the mitochondrial phenotype in fibroblasts isolated from airway biopsies of non-asthmatic and severe asthmatic subjects by examining mitophagy as a mechanism contributing to fibroblast persistence and thereby, fibrosis in severe asthma.
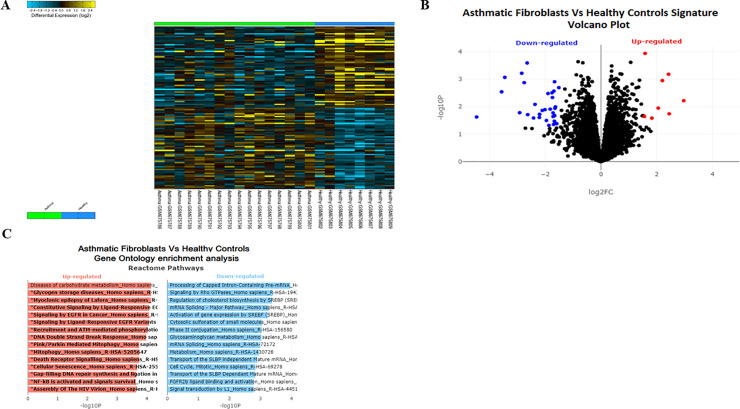
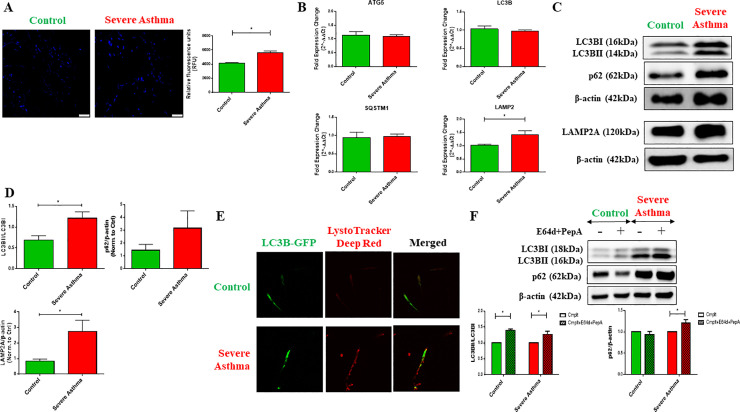
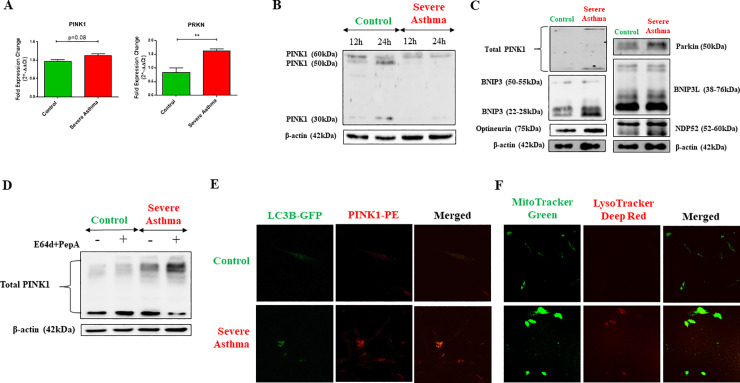
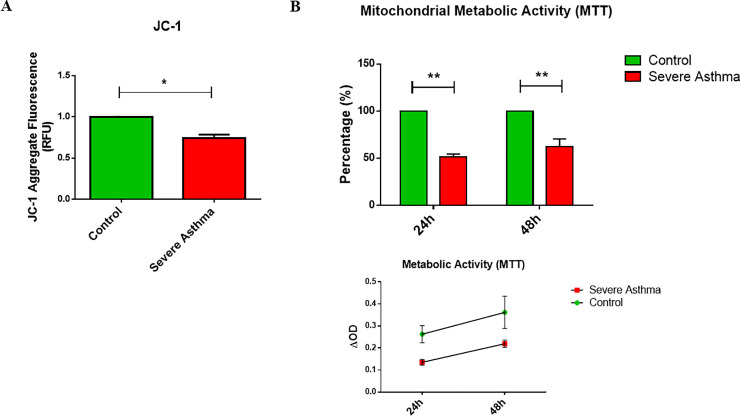
Methods: Bioinformatics analysis of publicly available transcriptomic data was performed to identify the top enriched pathways in asthmatic fibroblasts. Endogenous expression of mitophagy markers in severe asthmatic and non-asthmatic fibroblasts was determined using qRT-PCR, western blot and immunofluorescence. Mitophagy flux was examined by using lysosomal protease inhibitors, E64d and pepstatin A. Mitochondrial membrane potential and metabolic activity were also evaluated using JC-1 assay and MTT assay, respectively.
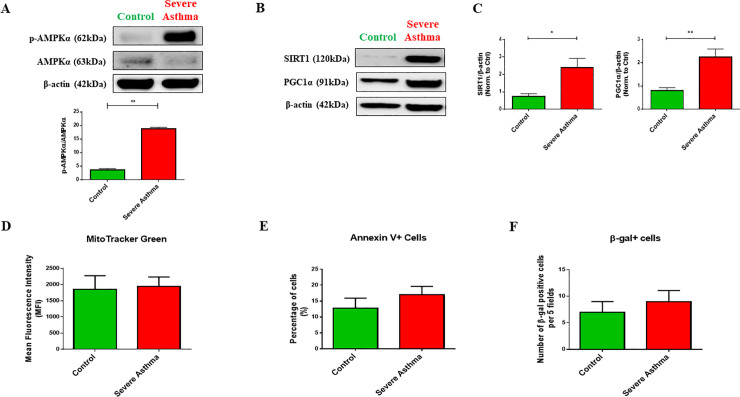
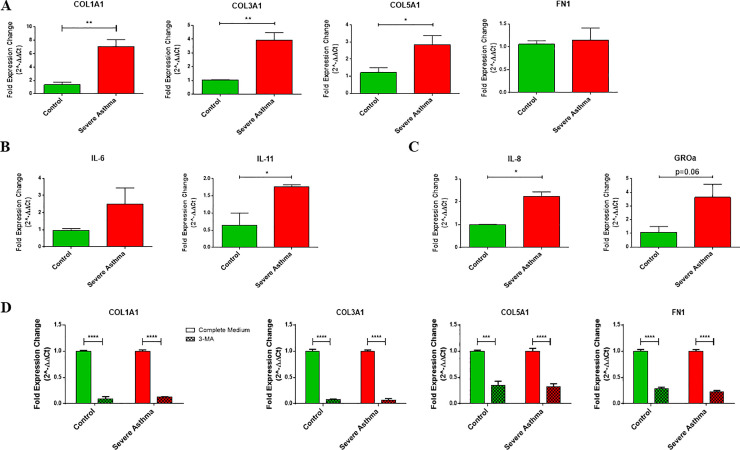
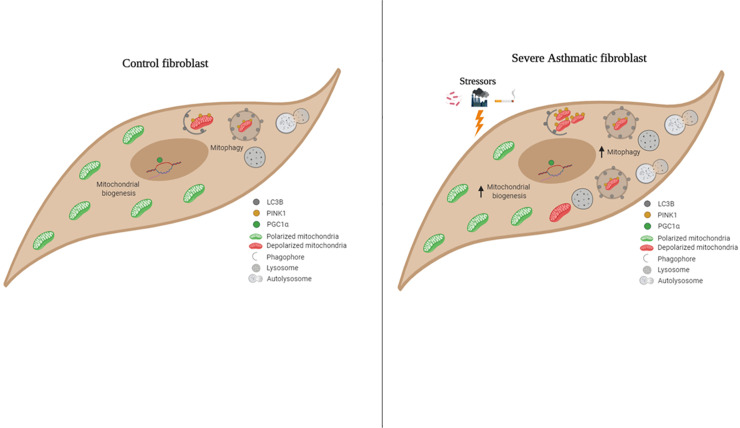
Results: Bioinformatics analysis revealed the enrichment of Pink/Parkin-mediated mitophagy in asthmatic fibroblasts compared to healthy controls. In severe asthmatic fibroblasts, the differential expression of mitophagy genes, PINK1 and PRKN, was accompanied by the accumulation of PINK1, Parkin and other mitophagy proteins at baseline. The further accumulation of endogenous LC3BII, p62 and PINK1 in the presence of E64d and pepstatin A in severe asthmatic fibroblasts reinforced their enhanced mitophagy flux. Significantly reduced mitochondrial membrane potential and metabolic activity were also demonstrated at baseline confirming the impairment in mitochondrial function in severe asthmatic fibroblasts. Interestingly, these fibroblasts displayed neither an apoptotic nor senescent phenotype but a pro-fibrotic phenotype with an adaptive survival mechanism triggered by increased AMPKα phosphorylation and mitochondrial biogenesis.
Conclusions: Our results demonstrated a role for mitophagy in the pathogenesis of severe asthma where the enhanced turnover of damaged mitochondria may contribute to fibrosis in severe asthma by promoting the persistence and pro-fibrotic phenotype of fibroblasts.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures