

Global discovery of human-infective RNA viruses: A modelling analysis
- PMID: 33253277
- PMCID: PMC7728385
- DOI: 10.1371/journal.ppat.1009079
Global discovery of human-infective RNA viruses: A modelling analysis
Abstract
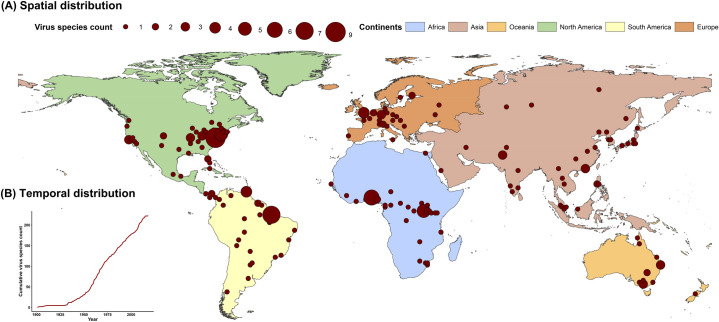
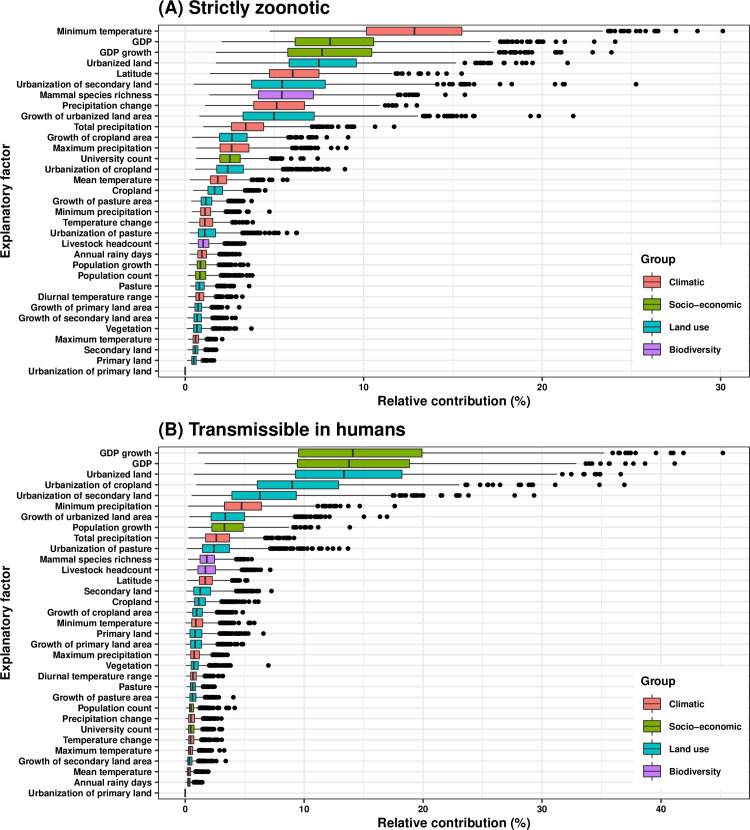
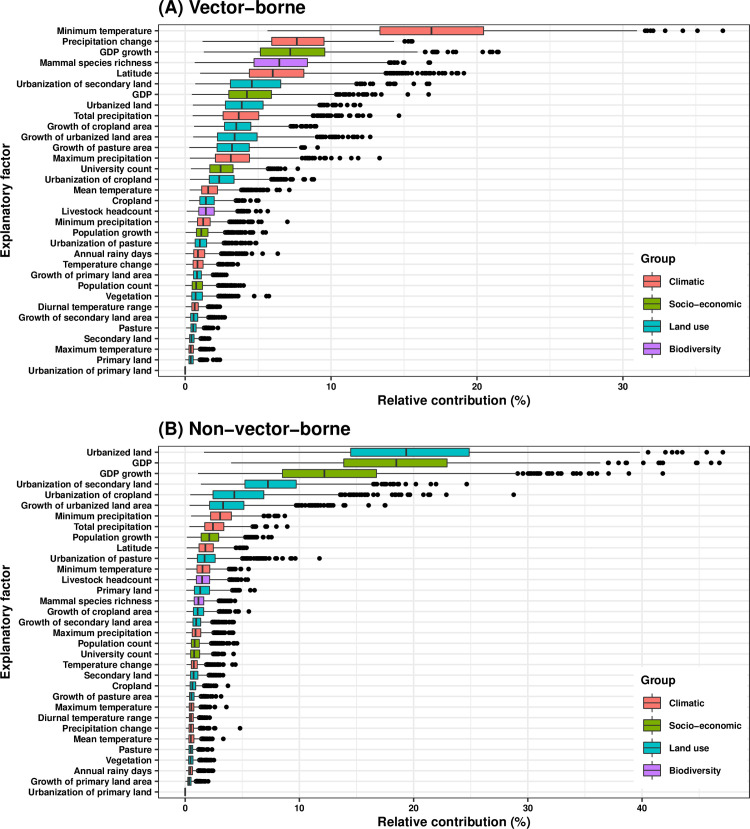
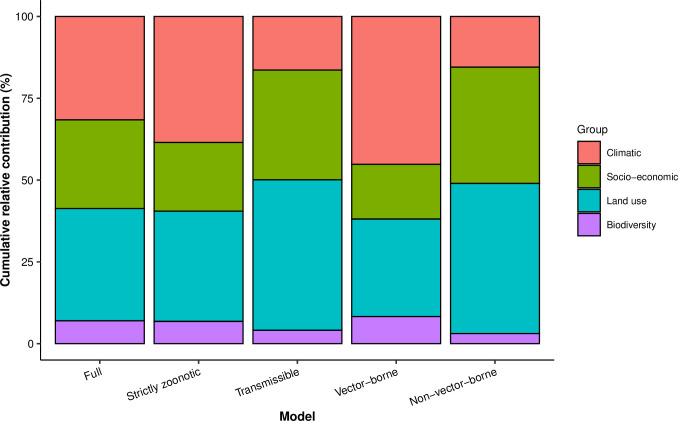
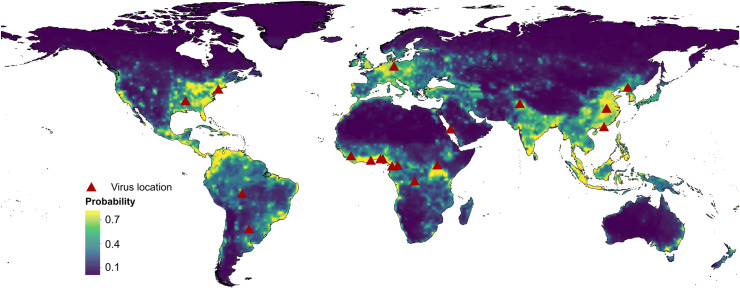
RNA viruses are a leading cause of human infectious diseases and the prediction of where new RNA viruses are likely to be discovered is a significant public health concern. Here, we geocoded the first peer-reviewed reports of 223 human RNA viruses. Using a boosted regression tree model, we matched these virus data with 33 explanatory factors related to natural virus distribution and research effort to predict the probability of virus discovery across the globe in 2010-2019. Stratified analyses by virus transmissibility and transmission mode were also performed. The historical discovery of human RNA viruses has been concentrated in eastern North America, Europe, central Africa, eastern Australia, and north-eastern South America. The virus discovery can be predicted by a combination of socio-economic, land use, climate, and biodiversity variables. Remarkably, vector-borne viruses and strictly zoonotic viruses are more associated with climate and biodiversity whereas non-vector-borne viruses and human transmissible viruses are more associated with GDP and urbanization. The areas with the highest predicted probability for 2010-2019 include three new regions including East and Southeast Asia, India, and Central America, which likely reflect both increasing surveillance and diversity of their virome. Our findings can inform priority regions for investment in surveillance systems for new human RNA viruses.
Conflict of interest statement
The authors disclose no conflicts of interest.
Figures

References
-
- WHO. HIV/AIDS report 2018 Geneva: World Health Organization; Available from: https://www.who.int/en/news-room/fact-sheets/detail/hiv-aids (accessed 19 July 2018).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
