

doi: 10.3324/haematol.2020.262006.
Eculizumab for complement mediated thrombotic microangiopathy in sickle cell disease
Affiliations
- PMID: 33256394
- PMCID: PMC7716365
- DOI: 10.3324/haematol.2020.262006
Item in Clipboard
Eculizumab for complement mediated thrombotic microangiopathy in sickle cell disease
Haematologica.
.
No abstract available
Figures
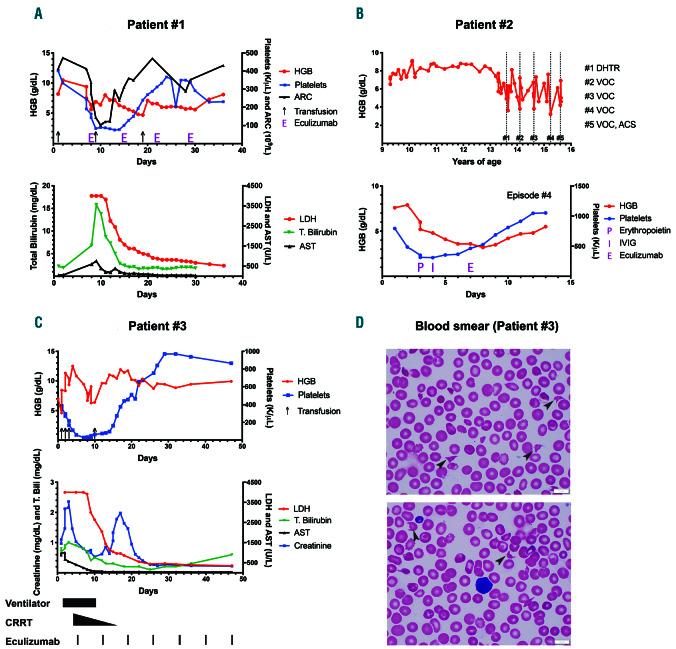
A graphic representation of a delayed hemolytic transfusion reaction, vaso-occlusive crisis and drug induced immune hemolytic anemia episode. (1A) DHTR (delayed hemolytic transfusion reaction): the patient received a transfusion (black arrows) on day one and presented with DHTR on day 7 with a hemoglobin (HGB) of 9.4 g/dL, which dropped to a nadir of 5.6 g/dL by day 8 along with absolute reticulocytopenia, as expected in DHTR. Thrombocytopenia coincided with this severe anemia and elevations in lactate dehydrogenase (LDH), total bilirubin (T.Bili), and aspartate transaminase (AST). Other evidence for intravascular hemolysis included reduced haptoglobin (<14; reference 30-120 mg/dL) and elevated plasma free HGB (120; reference < 30 mg/dL) not shown. Eculizumab (E) 600 mg was initiated on day 8 and given weekly for a total of four doses. The patient developed acute chest syndrome (ACS) and received one unit of crossmatch compatible, U-negative red blood cells (RBC) after a dose of intravenous immunoglobulin (IVIG) on day 9. The resolution of hemolysis was evidenced by improvement in LDH and HGB following the first dose of eculizumab and maintained throughout the hospital stay. Thrombocytopenia recovered within 1 week of complement inhibition. The patient required an additional transfusion on day 19 from rebound anemia likely secondary to frequent blood draws, since markers of hemolysis and complement activation on day 22 did not worsen. Immunohematology work-up during this DHTR episode: direct antiglobulin test (DAT) negative, historical anti-U detected. Note: the initial three LDH values were greater than 4,000 U/L (upper limit of our clinical laboratory detection). Eculizumab was dosed based on the weight of the patient, as per the guidelines for loading dose for children with atypical hemolytic uremic syndrome (aHUS). (1B) Vaso-occlusive crisis (VOC): grey colored dotted bars depict the time points when the patient presented with VOC and also corresponds to complement testing (numbered #1 through #5). Eculizumab was administered for episodes #3 and #4 as described. Lower graph represents episode #4 of VOC and shows an initial drop in HGB and platelets by day 4, when erythropoietin (P) 210 IU/kg was commenced along with a dose of IVIG (I) 1 g/kg on days 4 and 5, respectively. Given the continued deterioration in HGB to a nadir of 3.6 g/dL along with severe headache and worsening hypoxia, eculizumab (E) 900 mg was administered on day 7 after the blood for complement function analyses was collected. Rapid improvement in hemolysis and clinical status was observed within 48 hours of a single eculizumab dose. Eculizumab was dosed based on the weight of the patient, as per the guidelines for loading dose for children with aHUS. (1C) Drug induced immune hemolytic anemia (DIIHA): the patient presented on day 0 to the hospital with fever and received ceftriaxone; HGB dropped within 3 hours to 4 g/dL. Black arrows on the graph denote blood transfusions. Additional laboratory workup showed evidence of intravascular hemolysis, confirmed by elevated LDH >4,000 U/L, presence of schistocytes on blood smear and elevated plasma free HGB (not shown). Multiorgan failure was evident with a peak of creatinine at 2.36 mg/dL (baseline 0.3 mg/dL), and AST/alanine transaminase at 1,002/70 U/L along with rise in T.Bili. Black colored bars at the bottom of the graph depict the time points when various supportive care measures and eculizumab were administered. Shortly after the initiation of eculizumab, hemolysis decreased, as shown by the rapid drop in LDH, and the patient required less transfusion support. Thrombocytopenia improved. He had initial improvement in creatinine on continuous renal replacement therapy (CRRT), with a brief increase when CRRT was weaned. This rise was not sustained, and creatinine levels decreased promptly without any additional intervention except continued eculizumab therapy. By day 25, blood counts and chemistry were within normal limits. The rebound thrombocytosis persisted for few weeks before trending back to the patient’s baseline. Note: the initial three LDH values were greater than 4,000 U/L (upper limit of our clinical laboratory detection). Eculizumab was dosed based on the weight of the patient, as per the guidelines for loading dose for children with aHUS. Blood smears from days 4 and 5 showing the presence of schistocytes and helmet cells (black arrows), along with paucity of platelets. ARC: absolute reticulocyte count.
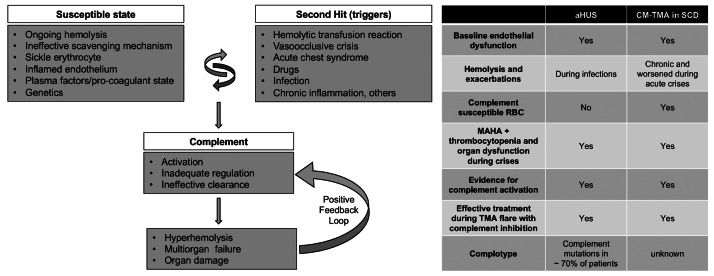
A model for complement- mediated thrombotic microangiopathy in sickle cell disease. Increased understanding of complement-mediated conditions such as atypical hemolytic uremic syndrome (aHUS) and paroxysmal nocturnal hemoglobinuria has renewed interest in understanding the specific role of complement in hyperhemolysis and innate immunity. Sickle cell disease (SCD) is a prototypical disease of chronic hemolysis, where increased levels of free plasma hemoglobin and heme with an insufficient, and thus ineffective scavenging mechanism by haptoglobin and hemopexin, leads to a state that is primed for complement activation. In addition, sickle erythrocytes themselves appear to be uniquely susceptible to complement-induced hemolysis, thereby further amplifying complement activation and additional hemolysis. Like in aHUS, the already inflamed endothelium in patients with SCD can be further modulated from increased hemolysis, complement activation, coagulation dysfunction, and other plasma proteins. Further genetic studies focusing on complotype are needed to help understand its role in SCD, as they can modulate the homeostatic balance of complement activity. Triggers such as pain crisis, acute chest syndrome, infection, etc. can drive this vulnerable state very quickly into a complement activated state, which, if unregulated, can result in common and terminal complement pathway activation that can lead to catastrophic damage in end organs and even death. Irrespective of the instigating cause, once complement-mediated hyperhemolysis is set off, it could result in a positive feedback loop causing further complement activation and a precipitous drop in hemoglobin and risk of sudden death. The table on the right parallels the resting and enabling state seen in SCD to patients with aHUS. CM-TMA: complement-mediated thrombotic microangiopathy; MAHA: microangiopathic hemolytic anemia; RBC: red blood cell.
References
-
- Vidler JB, Gardner K, Amenyah K, Mijovic A, Thein SL. Delayed haemolytic transfusion reaction in adults with sickle cell disease: a 5-year experience. Br J Haematol. 2015;169(5):746-753. - PubMed
-
- Habibi A, Mekontso-Dessap A, Guillaud C, et al. Delayed hemolytic transfusion reaction in adult sickle-cell disease: presentations, outcomes, and treatments of 99 referral center episodes. Am J Hematol. 2016;91(10):989-994. - PubMed
-
- Riedl M, Fakhouri F, Le Quintrec M, et al. Spectrum of complementmediated thrombotic microangiopathies: pathogenetic insights identifying novel treatment approaches. Semin Thromb Hemost. 2014;40(4):444-464. - PubMed
-
- Le Clech A, Simon-Tillaux N, Provôt F, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019;95(6):1443-1452. - PubMed
-
- Frimat M, Tabarin F, Dimitrov JD, et al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. 2013;122(2):282-292. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
