

Blood transcriptomics to characterize key biological pathways and identify biomarkers for predicting mortality in melioidosis
- PMID: 33256556
- PMCID: PMC7832033
- DOI: 10.1080/22221751.2020.1858176
Blood transcriptomics to characterize key biological pathways and identify biomarkers for predicting mortality in melioidosis
Abstract
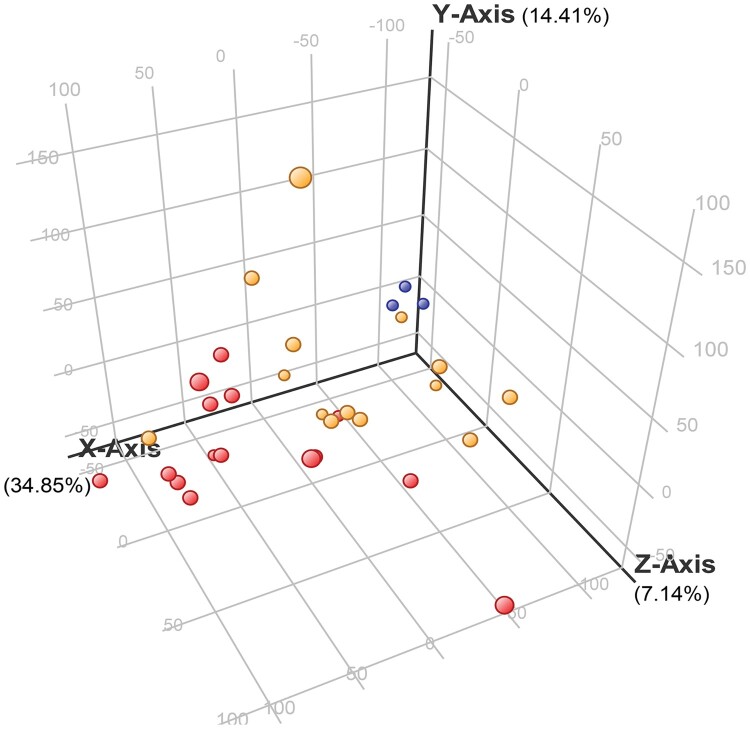
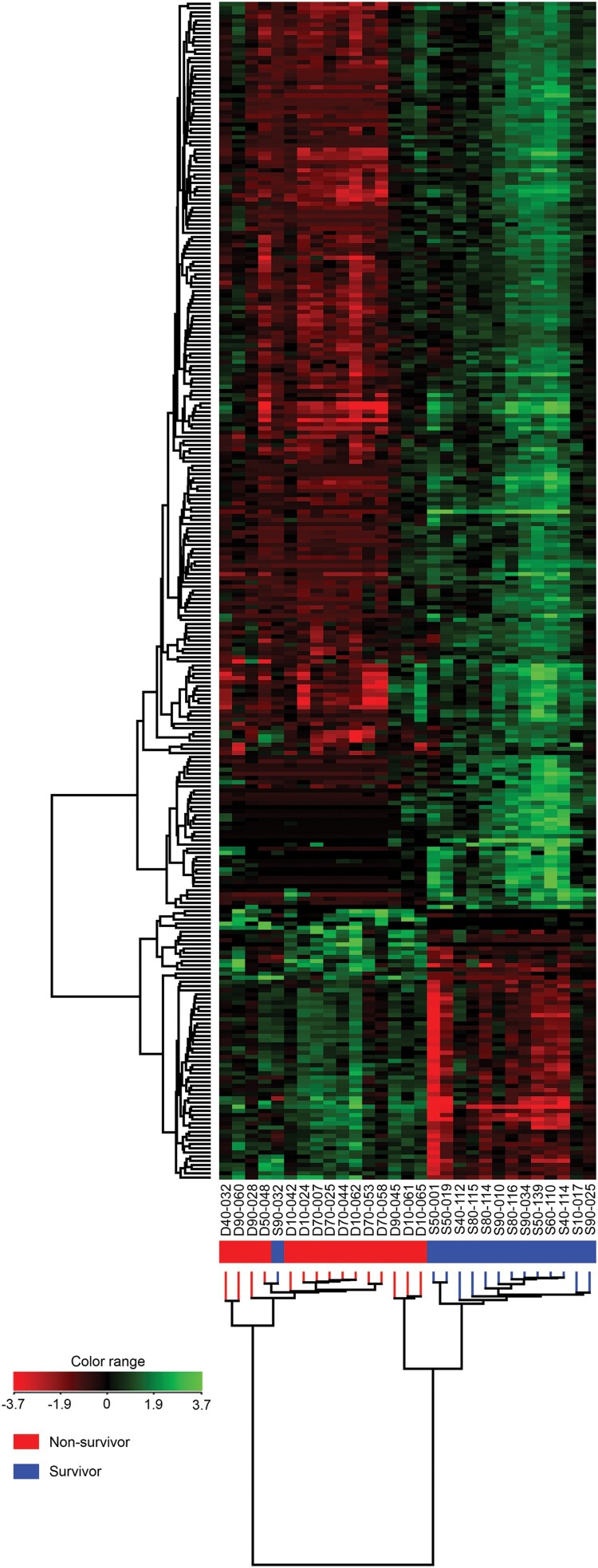
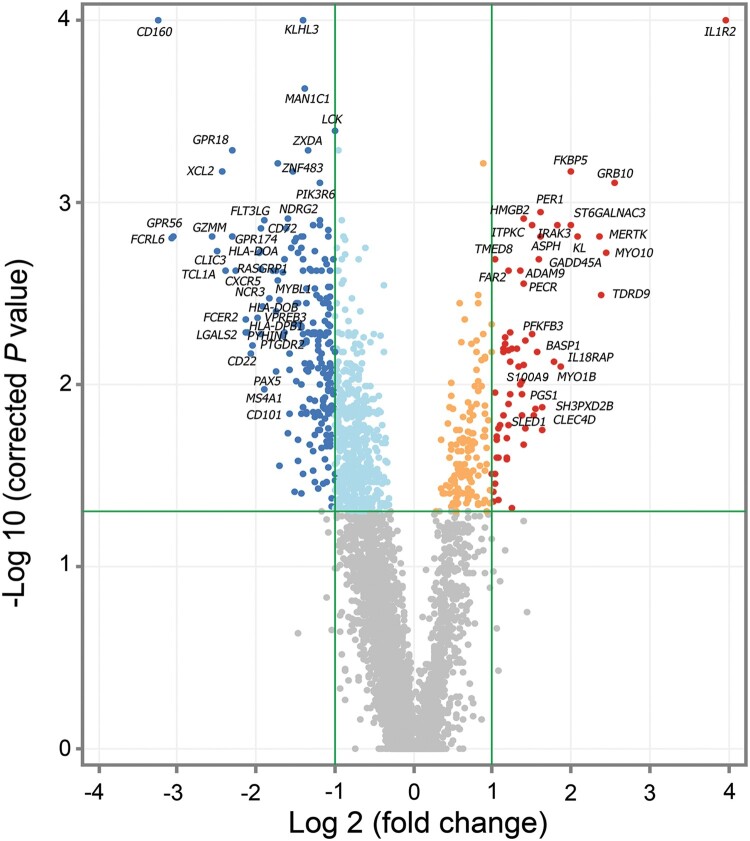
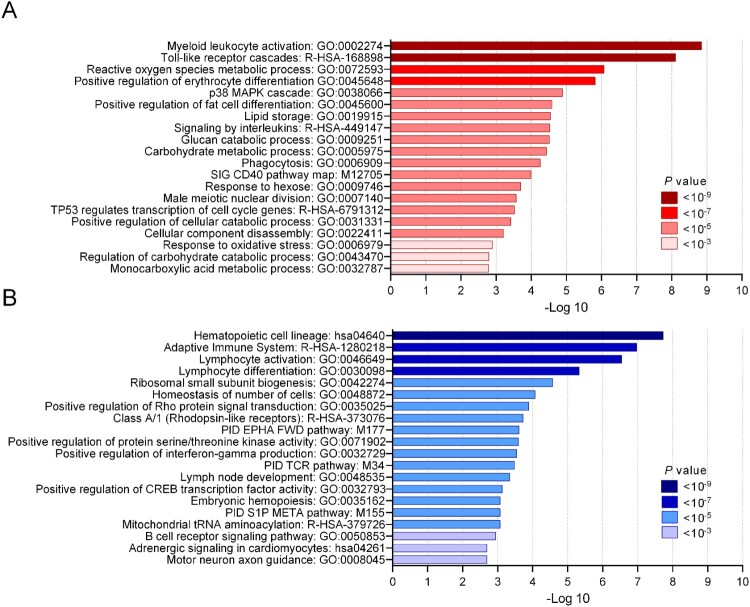
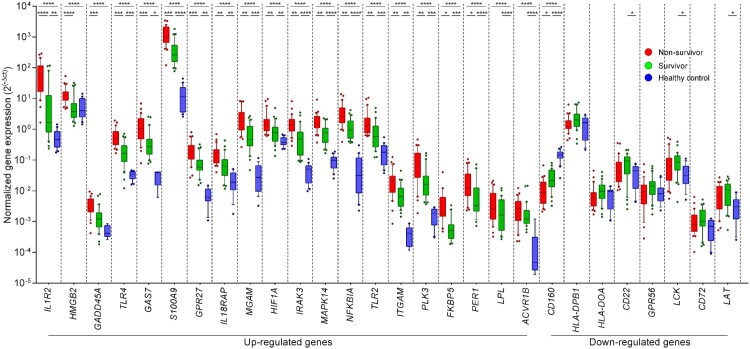
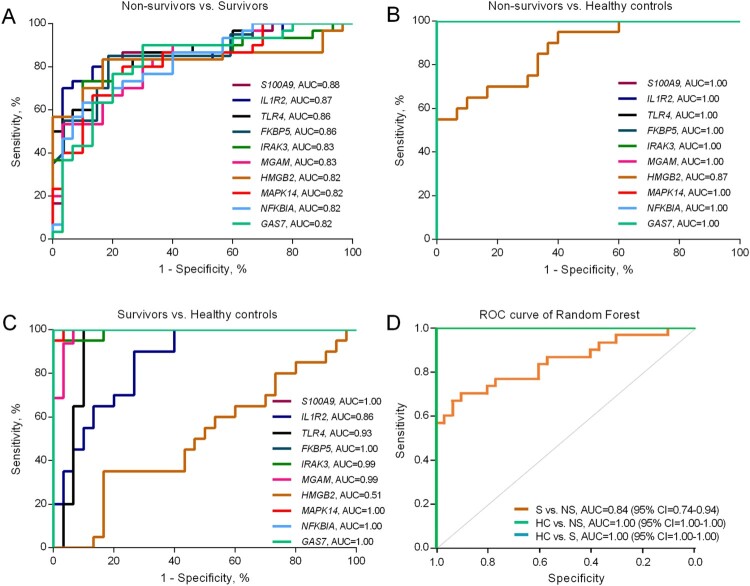
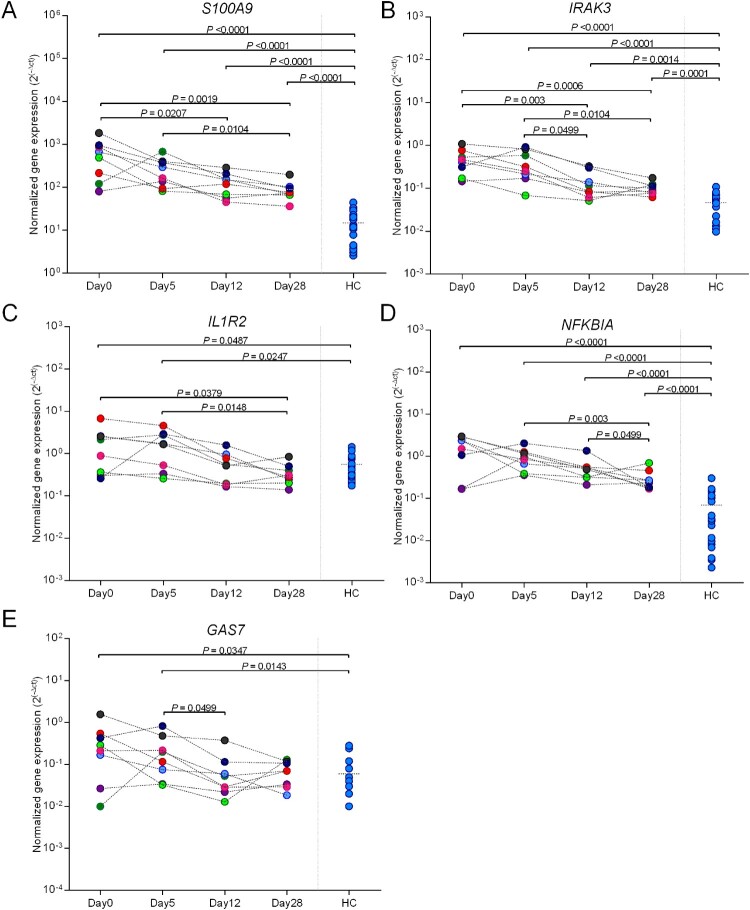
Melioidosis is an often lethal tropical disease caused by the Gram-negative bacillus, Burkholderia pseudomallei. The study objective was to characterize transcriptomes in melioidosis patients and identify genes associated with outcome. Whole blood RNA-seq was performed in a discovery set of 29 melioidosis patients and 3 healthy controls. Transcriptomic profiles of patients who did not survive to 28 days were compared with patients who survived and healthy controls, showing 65 genes were significantly up-regulated and 218 were down-regulated in non-survivors compared to survivors. Up-regulated genes were involved in myeloid leukocyte activation, Toll-like receptor cascades and reactive oxygen species metabolic processes. Down-regulated genes were hematopoietic cell lineage, adaptive immune system and lymphocyte activation pathways. RT-qPCR was performed for 28 genes in a validation set of 60 melioidosis patients and 20 healthy controls, confirming differential expression. IL1R2, GAS7, S100A9, IRAK3, and NFKBIA were significantly higher in non-survivors compared with survivors (P < 0.005) and healthy controls (P < 0.0001). The AUROCC of these genes for mortality discrimination ranged from 0.80-0.88. In survivors, expression of IL1R2, S100A9 and IRAK3 genes decreased significantly over 28 days (P < 0.05). These findings augment our understanding of this severe infection, showing expression levels of specific genes are potential biomarkers to predict melioidosis outcomes.
Keywords: Burkholderia pseudomallei; RNA-sequencing; biomarkers; immune response; melioidosis; outcome; transcriptomics.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous