

Pervasive generation of non-canonical subgenomic RNAs by SARS-CoV-2
- PMID: 33256807
- PMCID: PMC7704119
- DOI: 10.1186/s13073-020-00802-w
Pervasive generation of non-canonical subgenomic RNAs by SARS-CoV-2
Abstract
Background: SARS-CoV-2, a positive-sense RNA virus in the family Coronaviridae, has caused a worldwide pandemic of coronavirus disease 2019 or COVID-19. Coronaviruses generate a tiered series of subgenomic RNAs (sgRNAs) through a process involving homology between transcriptional regulatory sequences (TRS) located after the leader sequence in the 5' UTR (the TRS-L) and TRS located near the start of ORFs encoding structural and accessory proteins (TRS-B) near the 3' end of the genome. In addition to the canonical sgRNAs generated by SARS-CoV-2, non-canonical sgRNAs (nc-sgRNAs) have been reported. However, the consistency of these nc-sgRNAs across viral isolates and infection conditions is unknown. The comprehensive definition of SARS-CoV-2 RNA products is a key step in understanding SARS-CoV-2 pathogenesis.
Methods: Here, we report an integrative analysis of eight independent SARS-CoV-2 transcriptomes generated using three sequencing strategies, five host systems, and seven viral isolates. Read-mapping to the SARS-CoV-2 genome was used to determine the 5' and 3' coordinates of all junctions in viral RNAs identified in these samples.
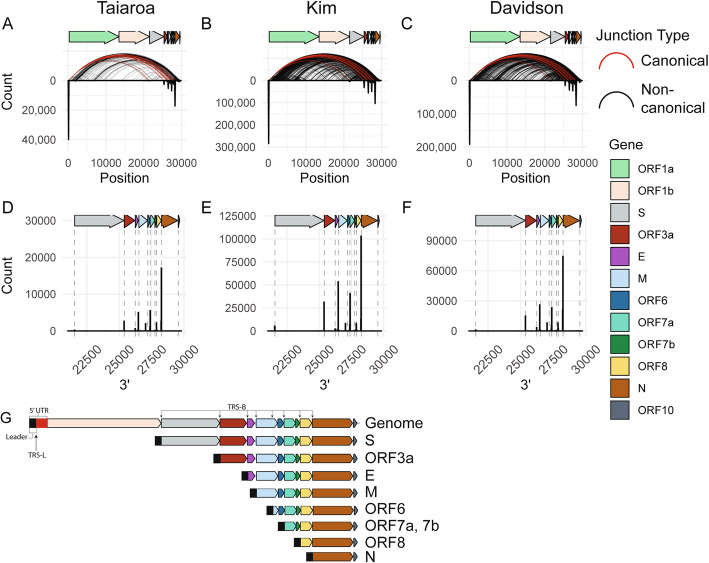
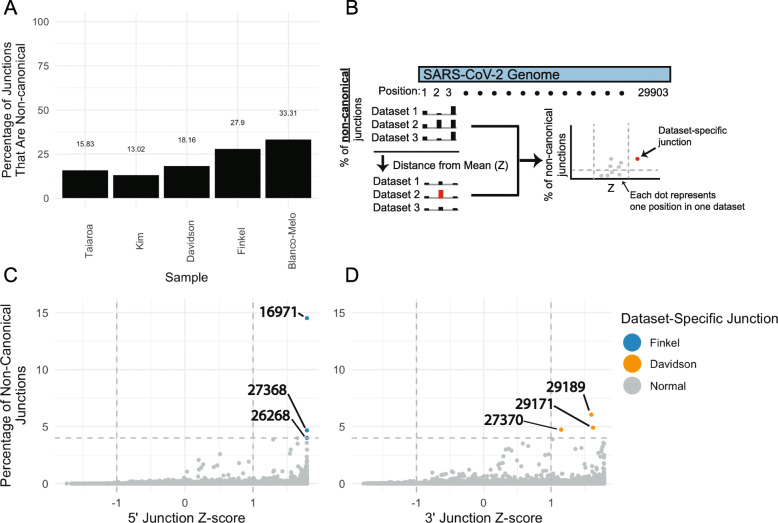
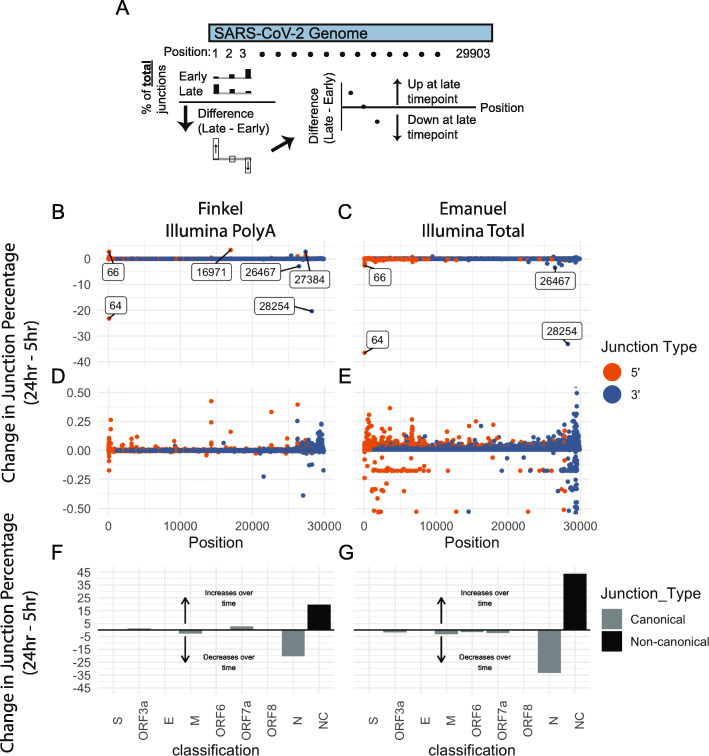
Results: Using junctional abundances, we show nc-sgRNAs make up as much as 33% of total sgRNAs in cell culture models of infection, are largely consistent in abundance across independent transcriptomes, and increase in abundance over time during infection. By assessing the homology between sequences flanking the 5' and 3' junction points, we show that nc-sgRNAs are not associated with TRS-like homology. By incorporating read coverage information, we find strong evidence for subgenomic RNAs that contain only 5' regions of ORF1a. Finally, we show that non-canonical junctions change the landscape of viral open reading frames.
Conclusions: We identify canonical and non-canonical junctions in SARS-CoV-2 sgRNAs and show that these RNA products are consistently generated by many independent viral isolates and sequencing approaches. These analyses highlight the diverse transcriptional activity of SARS-CoV-2 and offer important insights into SARS-CoV-2 biology.
Keywords: COVID-19; Direct RNA sequencing; SARS-CoV-2; Transcription.
Conflict of interest statement
M.M. receives research support from Bayer, Ono, and Janssen, has patents licensed to Bayer and Labcorp, and is a consultant for OrigiMed. J.A.D. received research support from Constellation Pharmaceuticals and is a consultant for EMD Serono, Inc. and Merck & Co. Inc. The remaining author declares no competing interests.
Figures



References
-
- Yount B, Roberts RS, Sims AC, Deming D, Frieman MB, Sparks J, et al. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J Virol. 2005;79(23):14909–14922. doi: 10.1128/JVI.79.23.14909-14922.2005. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
