

Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms
- PMID: 33257573
- PMCID: PMC7749287
- DOI: 10.1073/pnas.2005877117
Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms
Erratum in
-
Correction for Aras et al., Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) rescues the cellular phenotype of MELAS by inducing homeostatic mechanisms.Proc Natl Acad Sci U S A. 2021 Feb 16;118(7):e2100262118. doi: 10.1073/pnas.2100262118. Proc Natl Acad Sci U S A. 2021. PMID: 33526597 Free PMC article. No abstract available.
Abstract
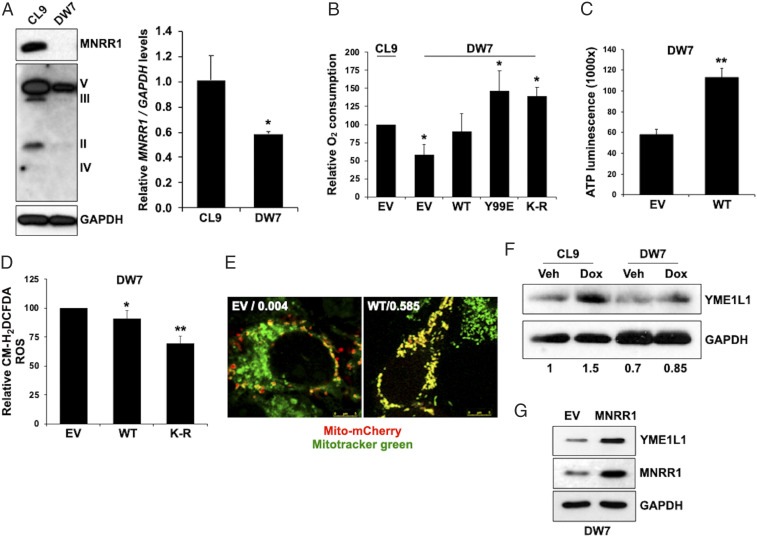
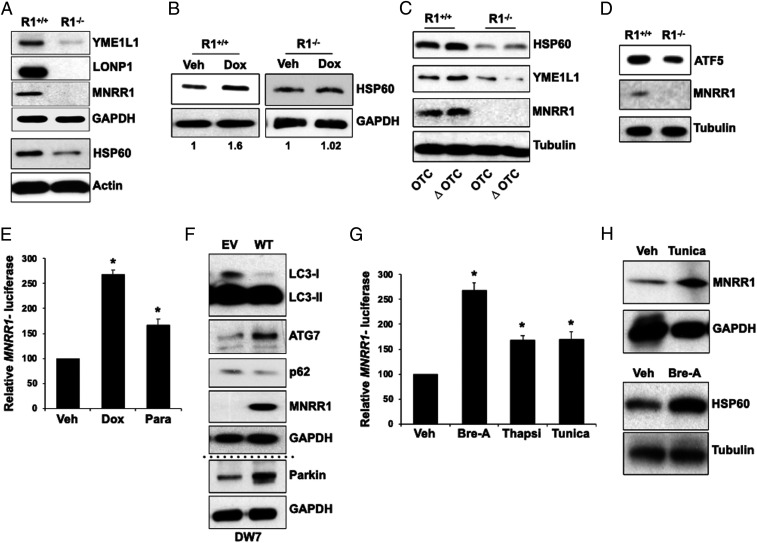
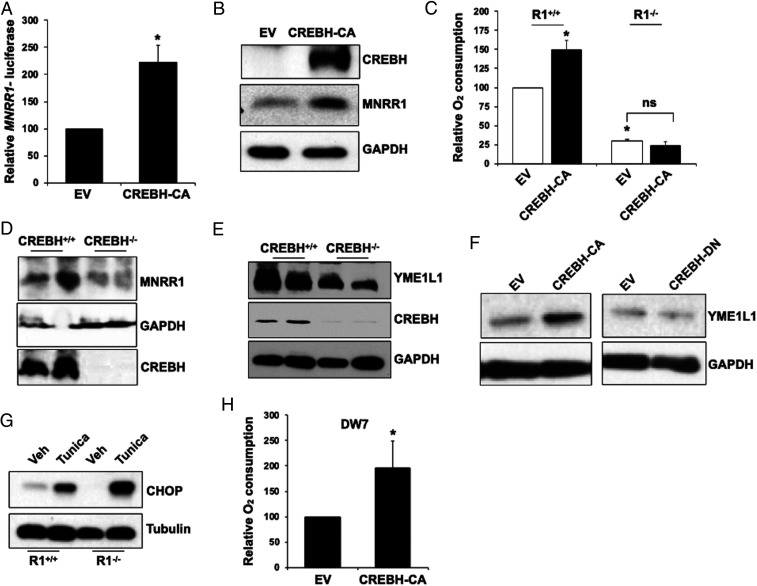
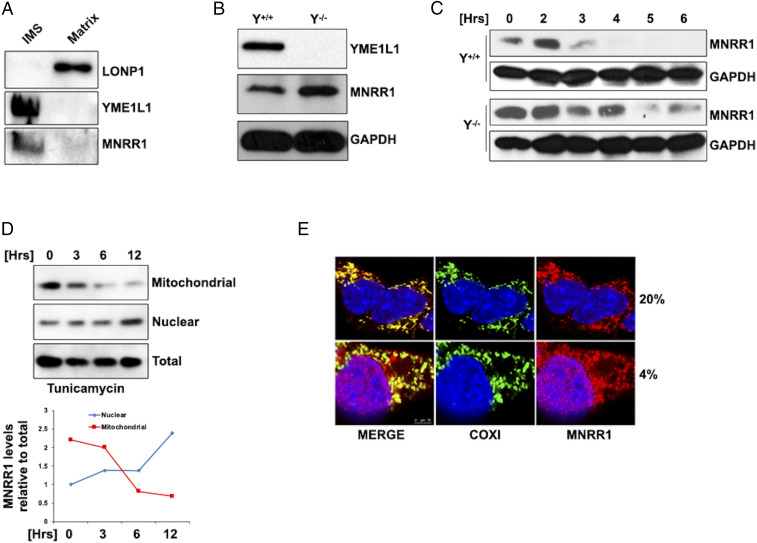
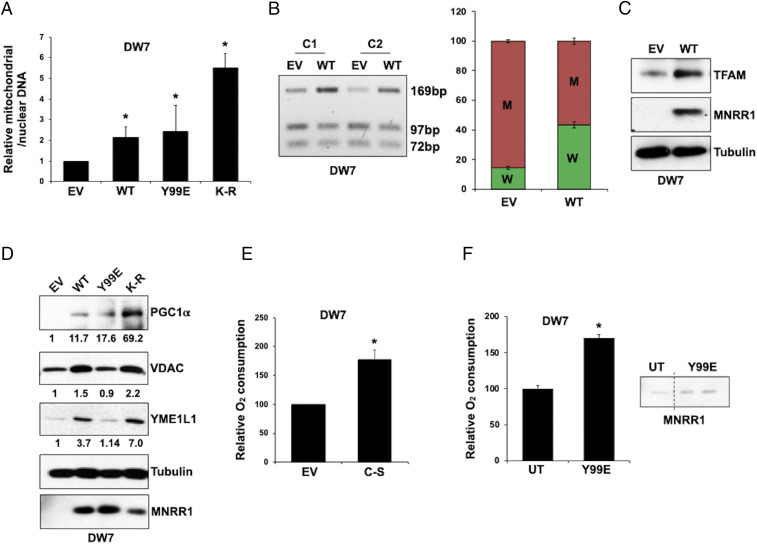
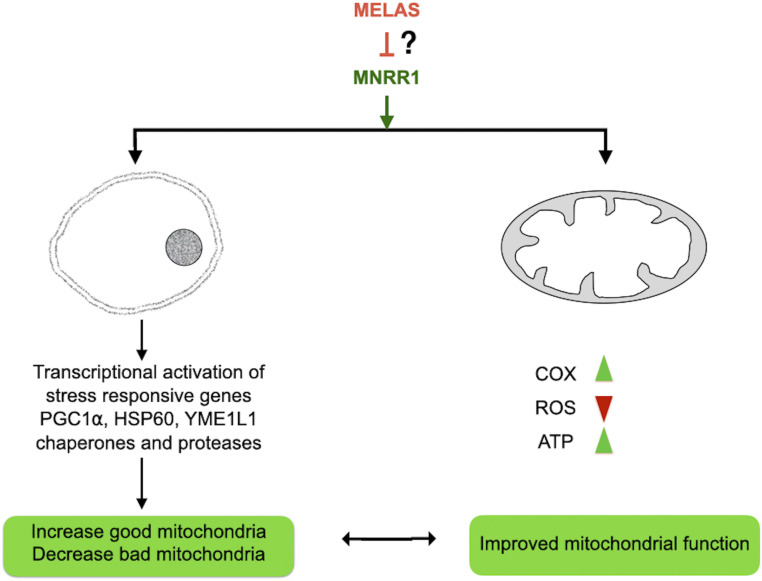
MNRR1 (CHCHD2) is a bi-organellar regulator of mitochondrial function that directly activates cytochrome c oxidase in the mitochondria and functions in the nucleus as a transcriptional activator for hundreds of genes. Since MNRR1 depletion contains features of a mitochondrial disease phenotype, we evaluated the effects of forced expression of MNRR1 on the mitochondrial disease MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes) syndrome. MELAS is a multisystem encephalomyopathy disorder that can result from a heteroplasmic mutation in the mitochondrial DNA (mtDNA; m.3243A > G) at heteroplasmy levels of ∼50 to 90%. Since cybrid cell lines with 73% m.3243A > G heteroplasmy (DW7) display a significant reduction in MNRR1 levels compared to the wild type (0% heteroplasmy) (CL9), we evaluated the effects of MNRR1 levels on mitochondrial functioning. Overexpression of MNRR1 in DW7 cells induces the mitochondrial unfolded protein response (UPRmt), autophagy, and mitochondrial biogenesis, thereby rescuing the mitochondrial phenotype. It does so primarily as a transcription activator, revealing this function to be a potential therapeutic target. The role of MNRR1 in stimulating UPRmt, which is blunted in MELAS cells, was surprising and further investigation uncovered that under conditions of stress the import of MNRR1 into the mitochondria was blocked, allowing the protein to accumulate in the nucleus to enhance its transcription function. In the mammalian system, ATF5, has been identified as a mediator of UPRmt MNRR1 knockout cells display an ∼40% reduction in the protein levels of ATF5, suggesting that MNRR1 plays an important role upstream of this known mediator of UPRmt.
Keywords: CHCHD2; cytochrome c oxidase; mitochondria; transcription; unfolded protein response.
Conflict of interest statement
Competing interest statement: Company associations for D.C.W. not related to the manuscript include MitoCURia, Mitrios, and Panos.
Figures
References
-
- Aras S., et al. , Abl2 kinase phosphorylates bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta Mol. Cell Res. 1864, 440–448 (2017). - PubMed
-
- Aras S., et al. , MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 20, 43–51 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
