

Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease
- PMID: 33257666
- PMCID: PMC7704703
- DOI: 10.1038/s41467-020-19737-2
Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease
Abstract
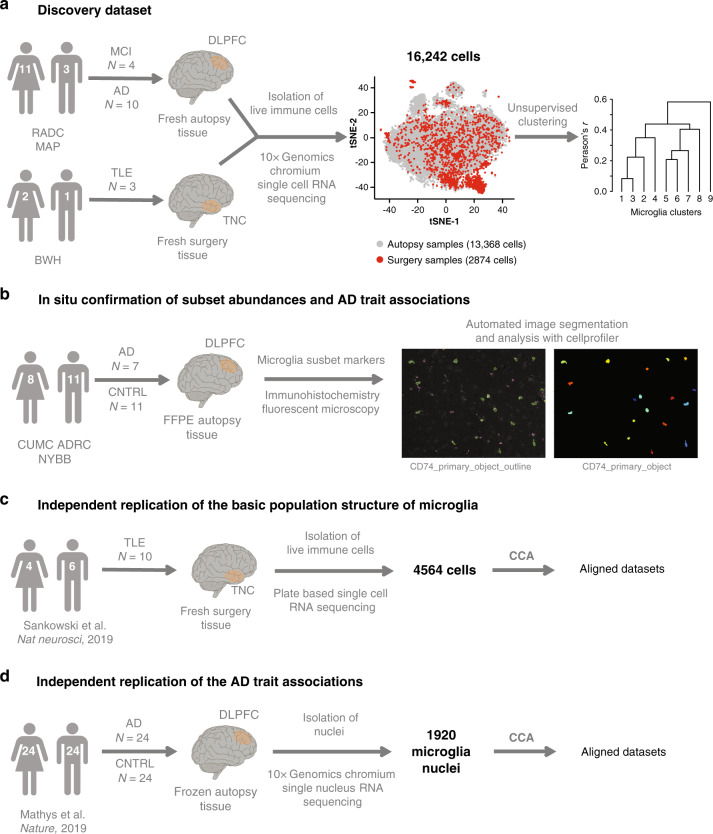
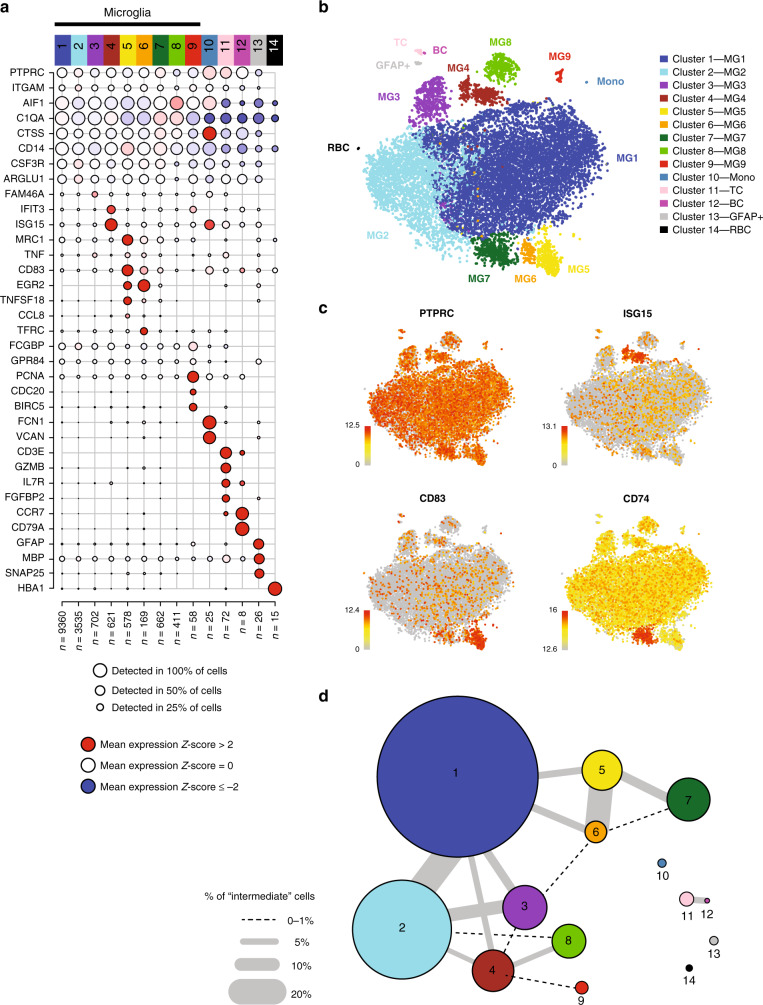
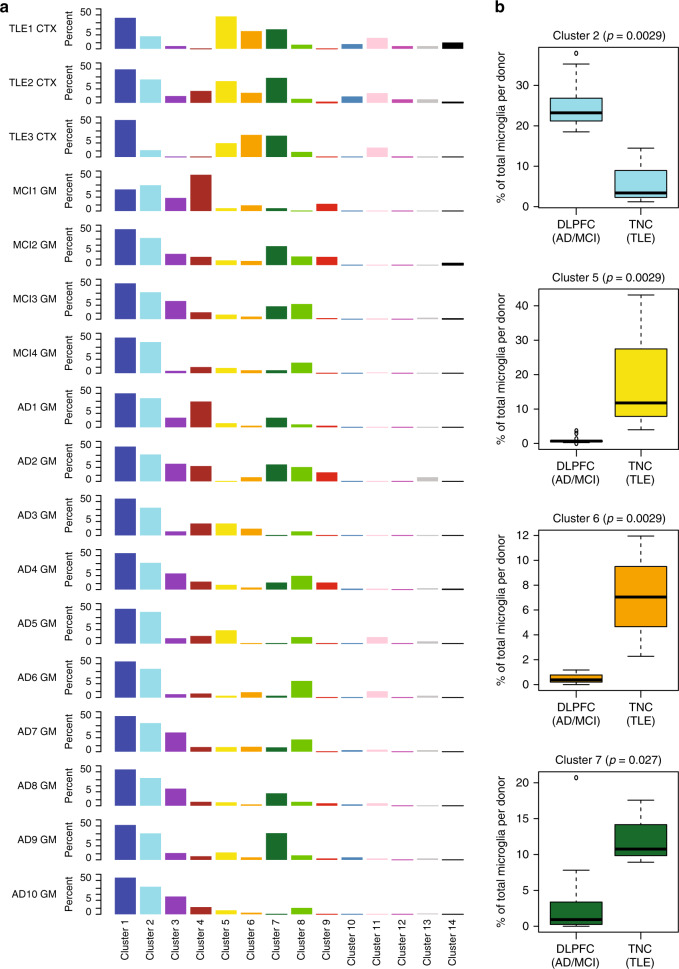
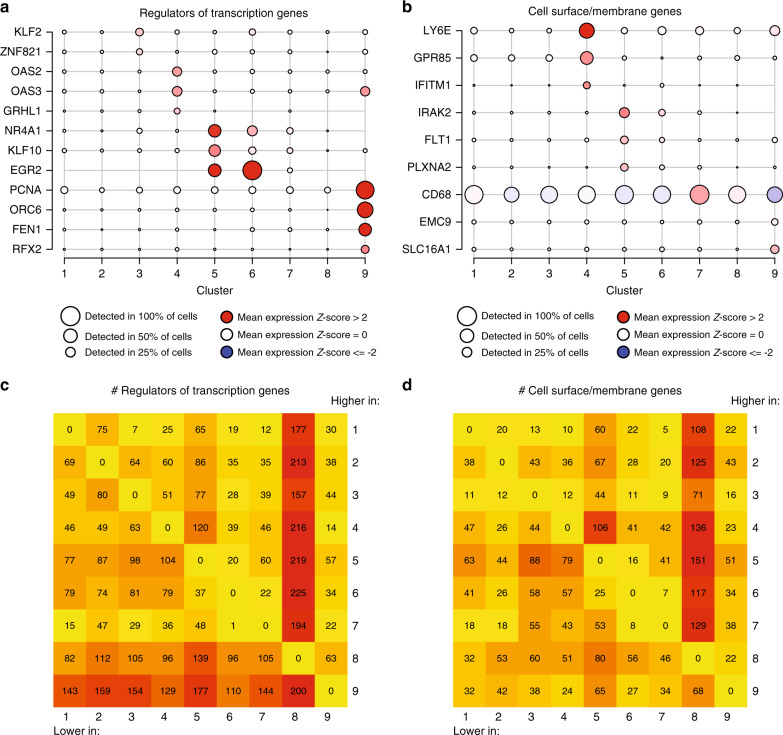
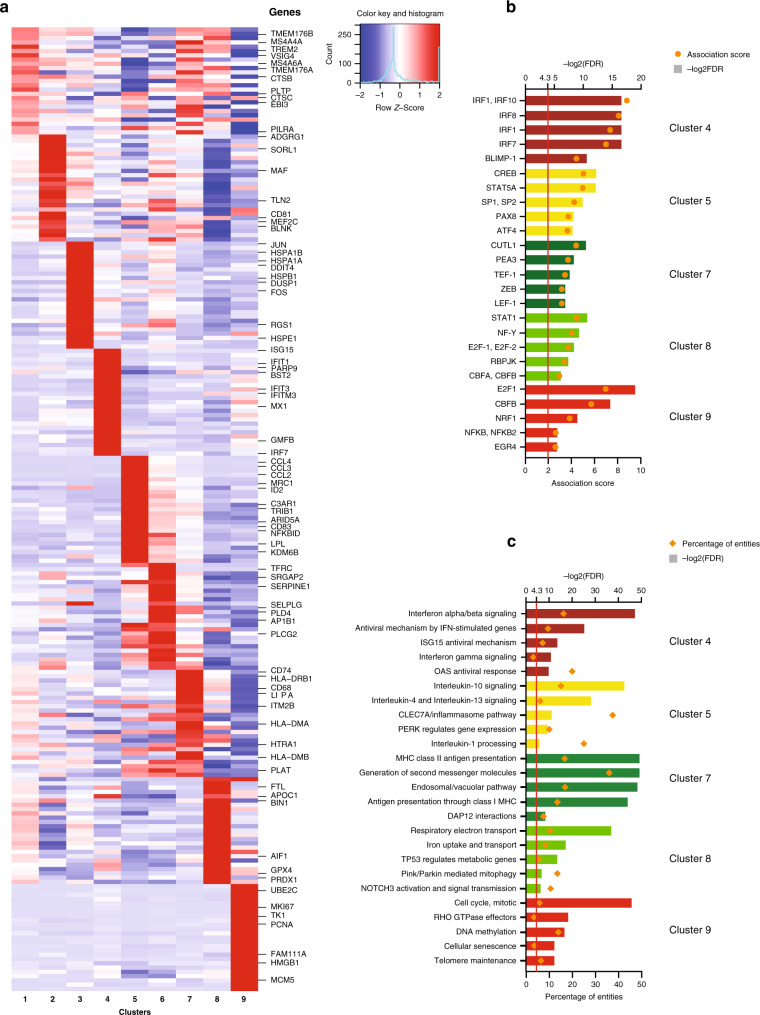
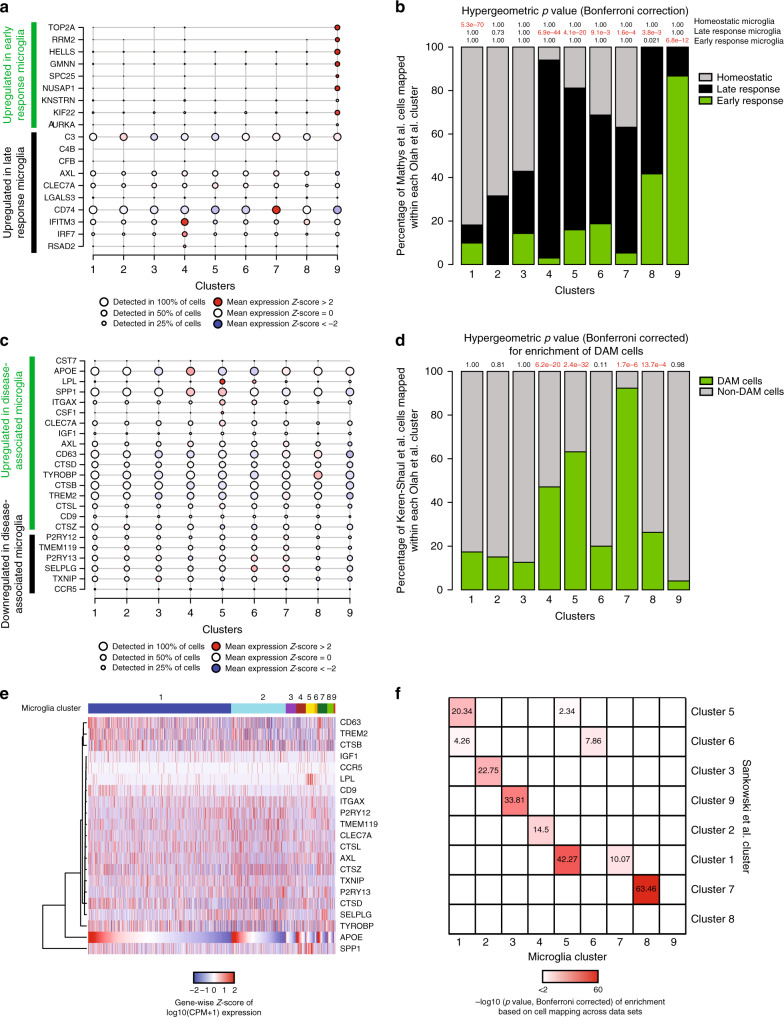
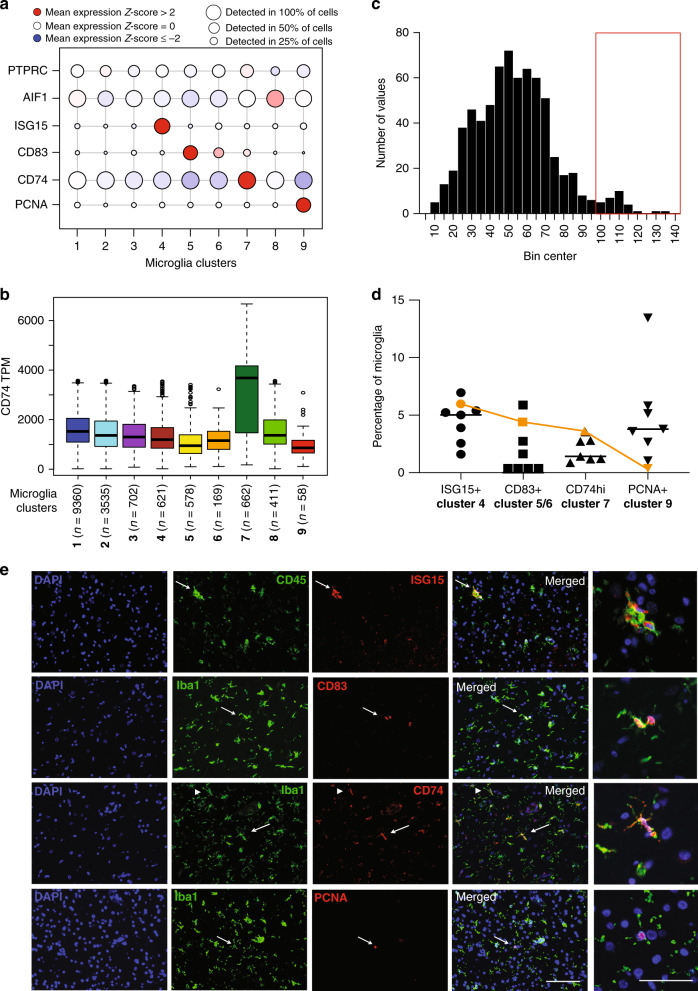
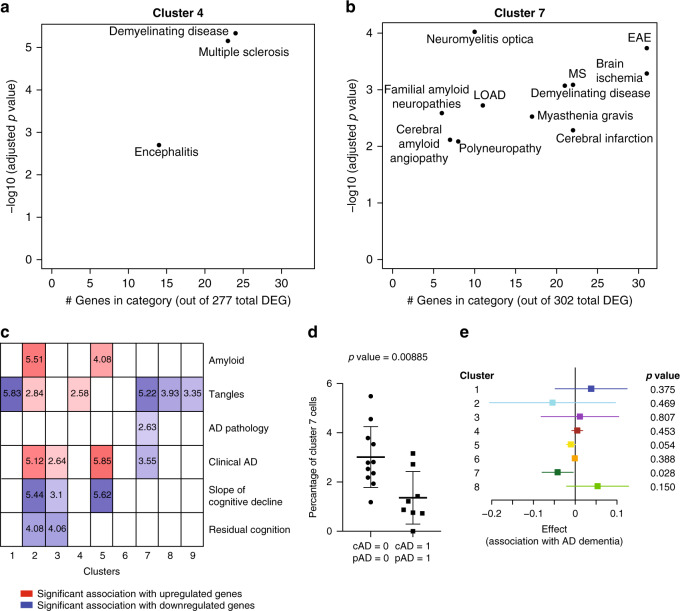
The extent of microglial heterogeneity in humans remains a central yet poorly explored question in light of the development of therapies targeting this cell type. Here, we investigate the population structure of live microglia purified from human cerebral cortex samples obtained at autopsy and during neurosurgical procedures. Using single cell RNA sequencing, we find that some subsets are enriched for disease-related genes and RNA signatures. We confirm the presence of four of these microglial subpopulations histologically and illustrate the utility of our data by characterizing further microglial cluster 7, enriched for genes depleted in the cortex of individuals with Alzheimer's disease (AD). Histologically, these cluster 7 microglia are reduced in frequency in AD tissue, and we validate this observation in an independent set of single nucleus data. Thus, our live human microglia identify a range of subtypes, and we prioritize one of these as being altered in AD.
Conflict of interest statement
A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas, and was an SAB member of ThermoFisher Scientific, Syros Pharmaceuticals, Neogene Therapeutics and Asimov. From August 1, 2020, A.R. is now an employee of Genentech. All other authors declare no competing interests.
Figures
References
-
- Stevens B., & Schafer D. P. Roles of microglia in nervous system development, plasticity, and disease. Dev. Neurobiol. 78, 559–560 (2018). - PubMed
Publication types
MeSH terms
Grants and funding
- R01 NS113516/NS/NINDS NIH HHS/United States
- U01 AG046152/AG/NIA NIH HHS/United States
- R56 NS089674/NS/NINDS NIH HHS/United States
- P50 AG008702/AG/NIA NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 AG017917/AG/NIA NIH HHS/United States
- P30 AG066462/AG/NIA NIH HHS/United States
- P30 AG010161/AG/NIA NIH HHS/United States
- RF1 AG058852/AG/NIA NIH HHS/United States
- RF1 AG057473/AG/NIA NIH HHS/United States
- R01 AG048015/AG/NIA NIH HHS/United States
- R01 NS089674/NS/NINDS NIH HHS/United States
- R01 AG036836/AG/NIA NIH HHS/United States
- R01 AG015819/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
