

Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED
- PMID: 33257863
- PMCID: PMC8567335
- DOI: 10.1038/s41587-020-0731-9
Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED
Abstract
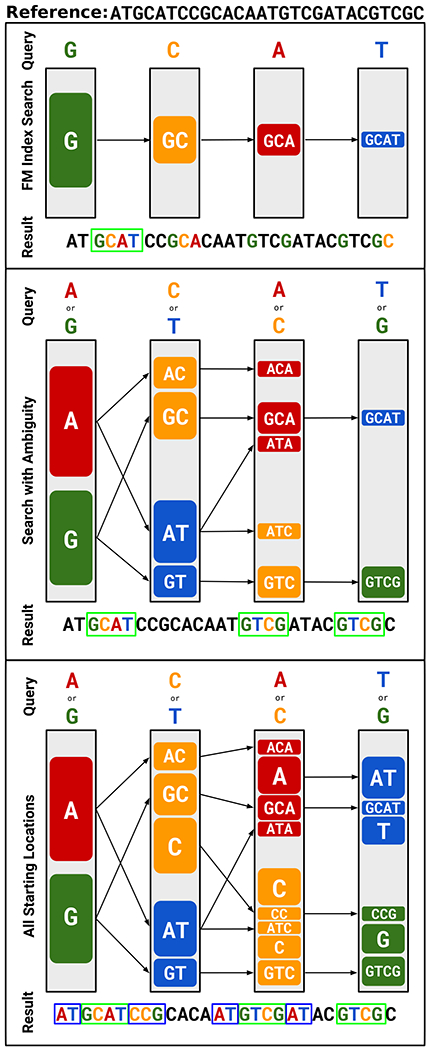
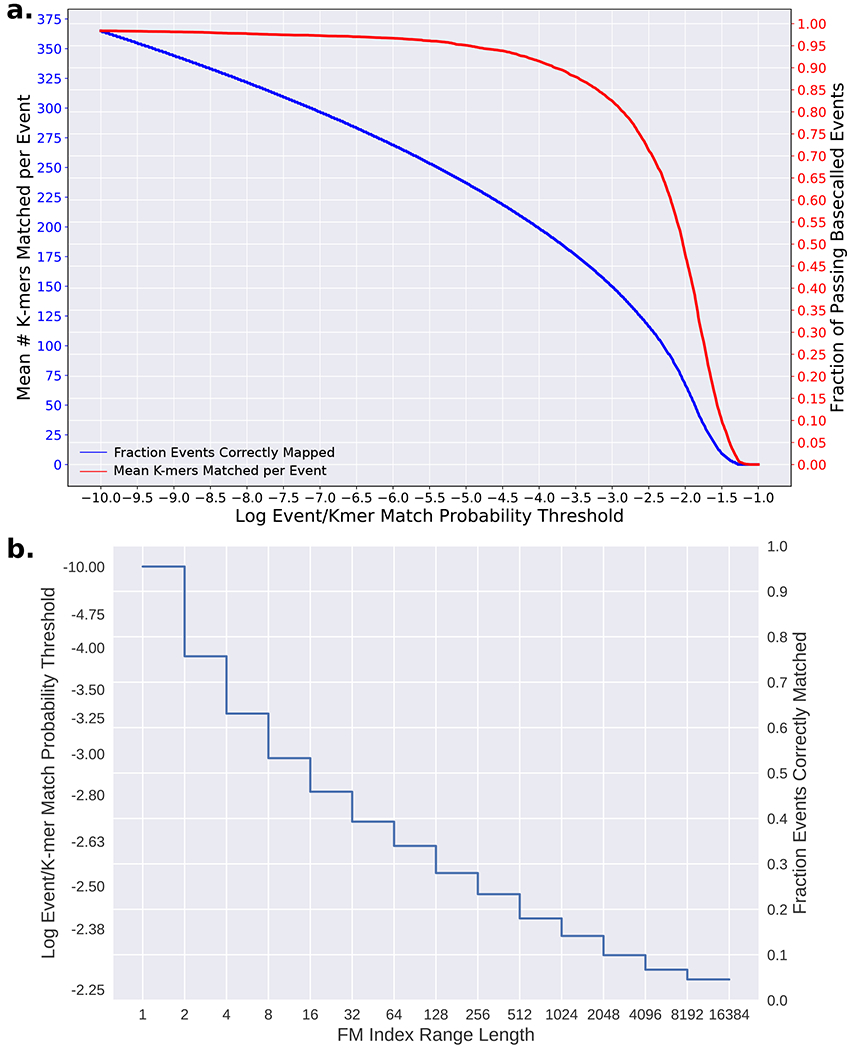
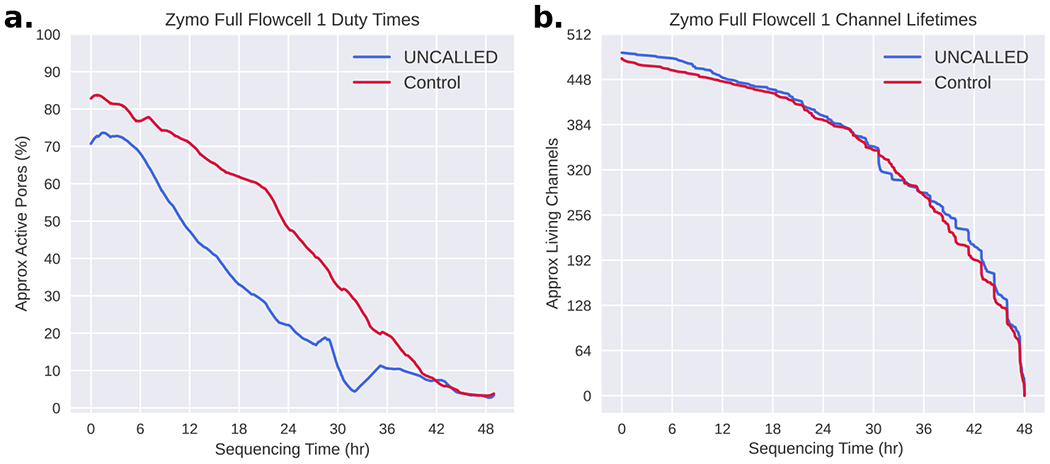
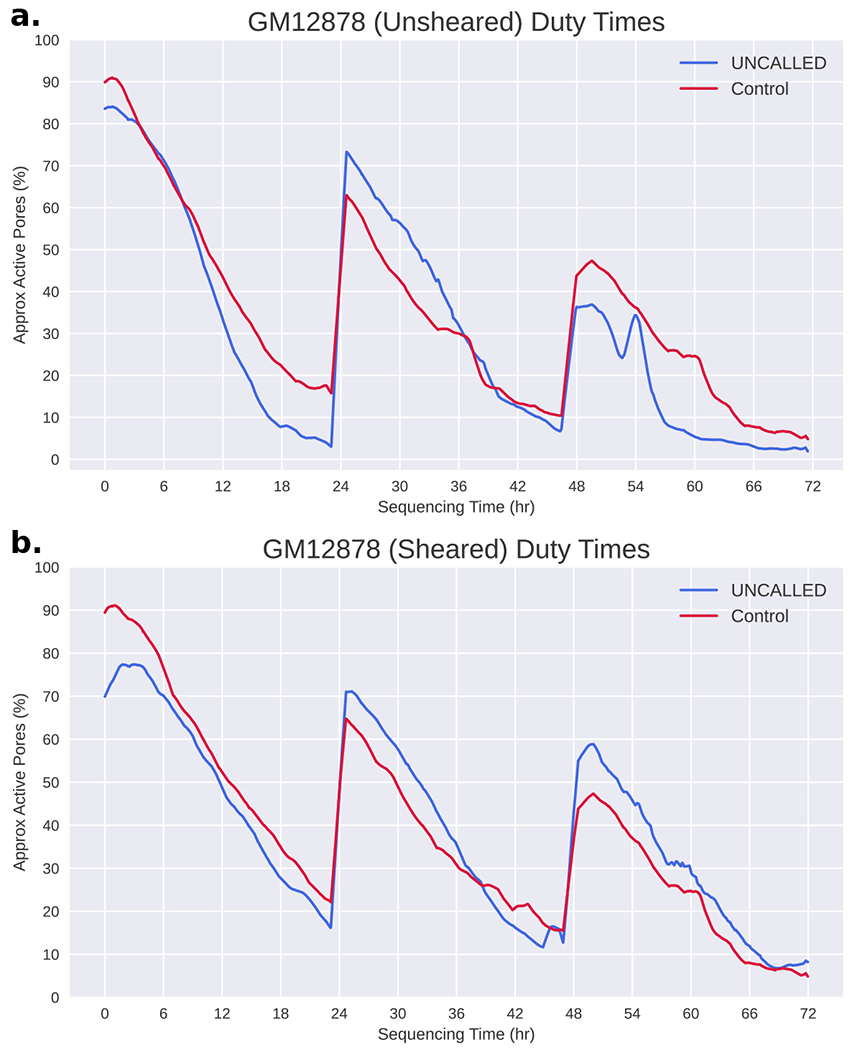
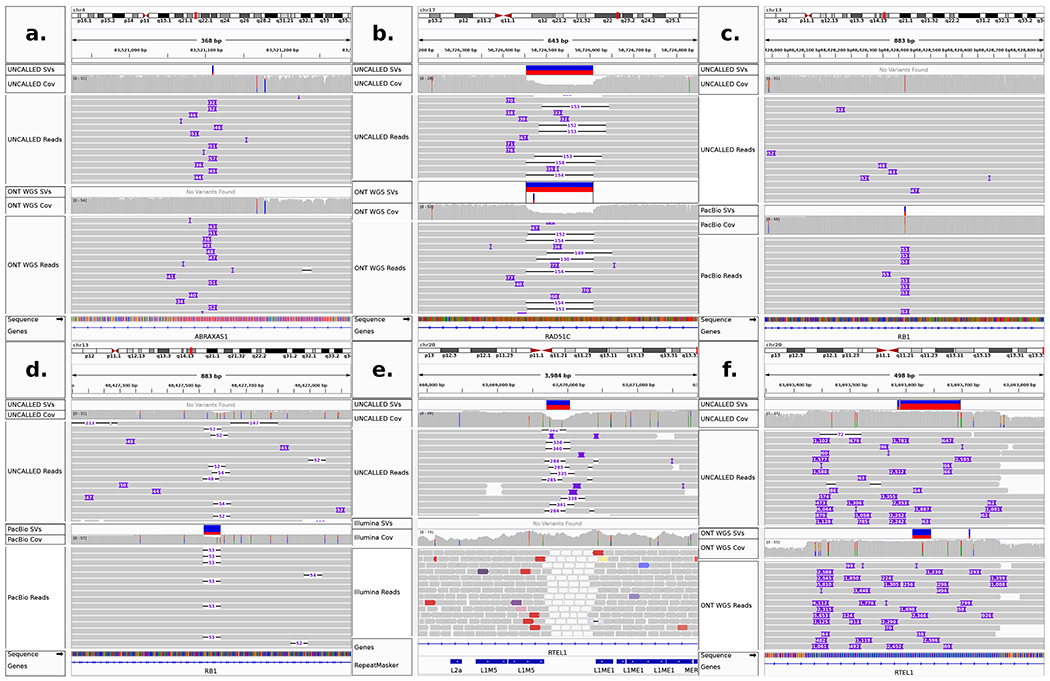
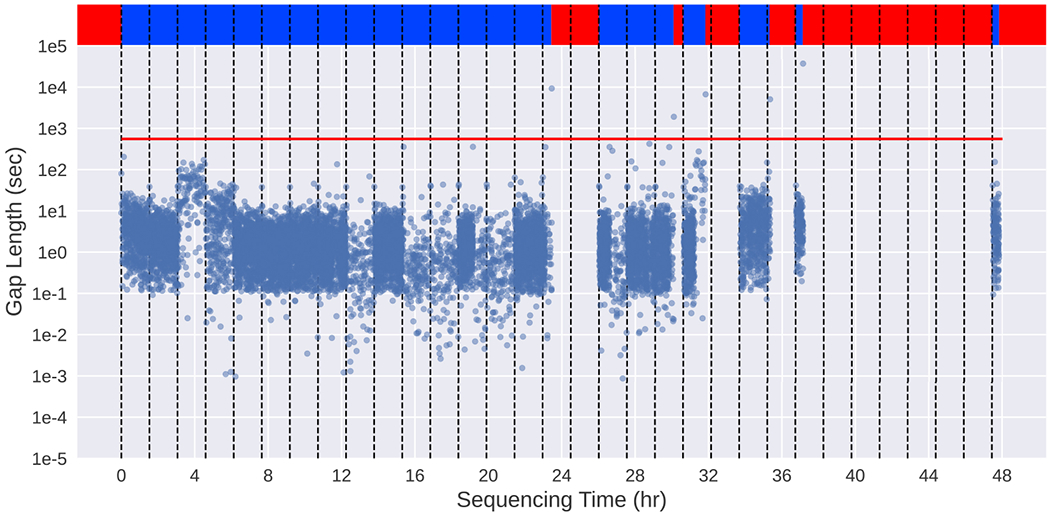
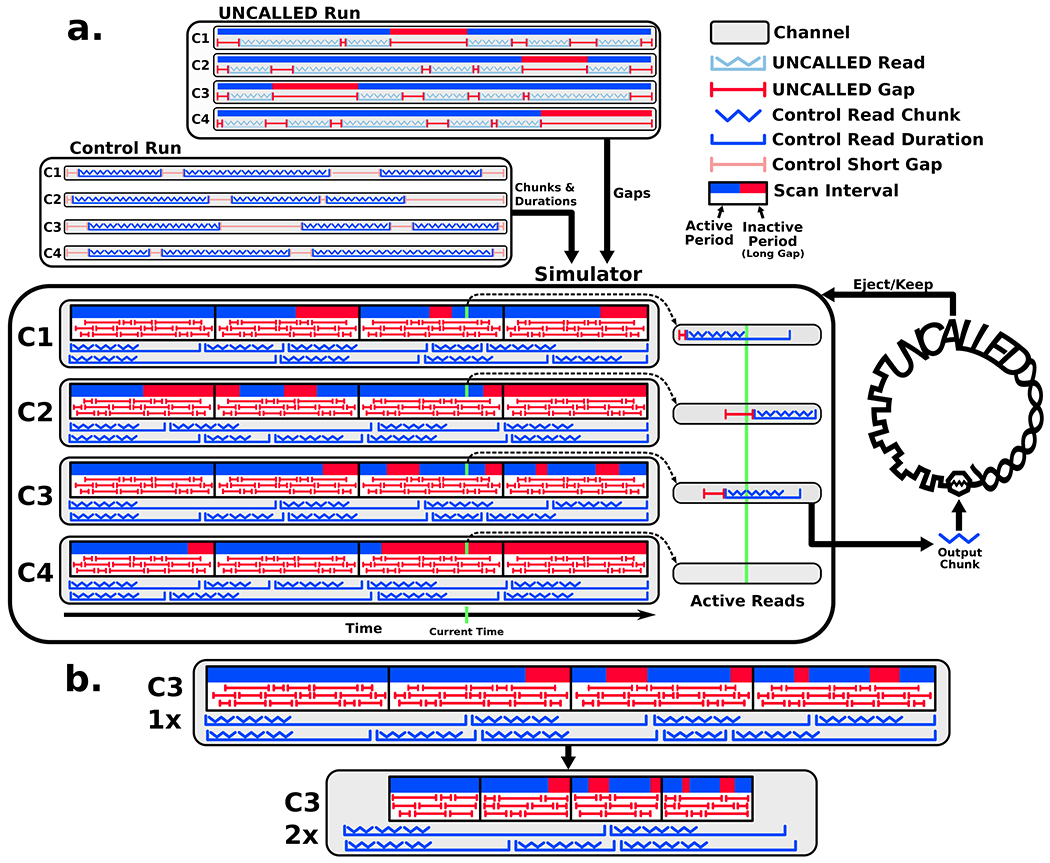
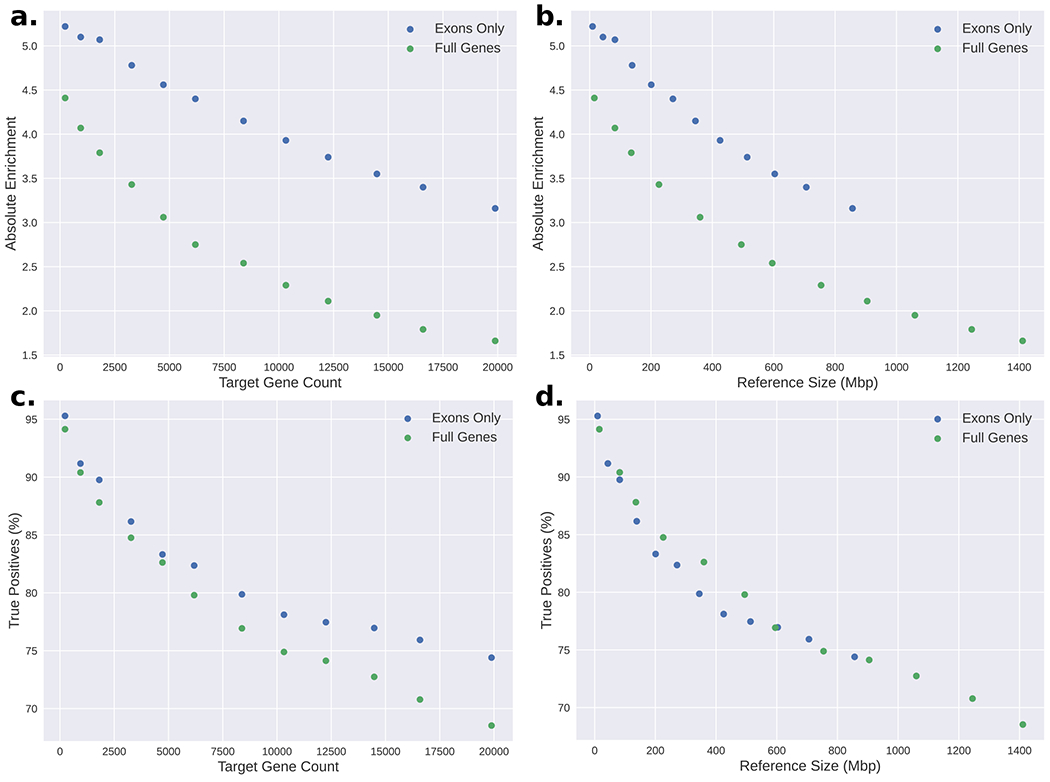
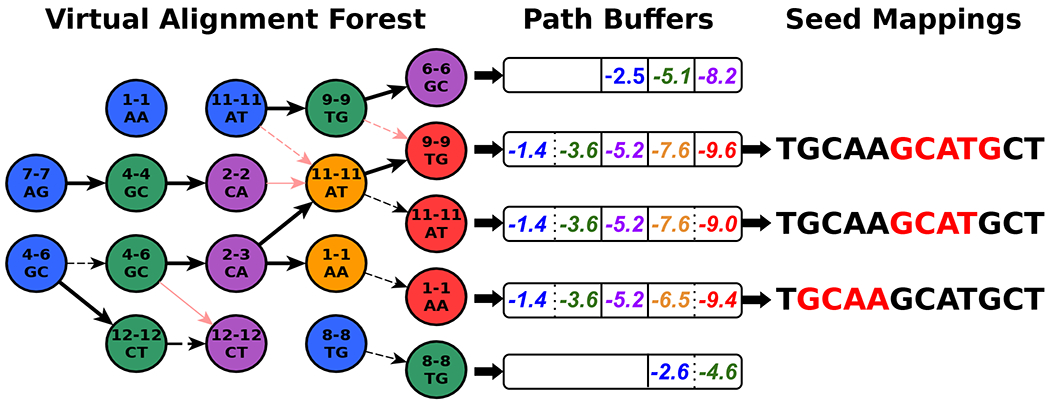
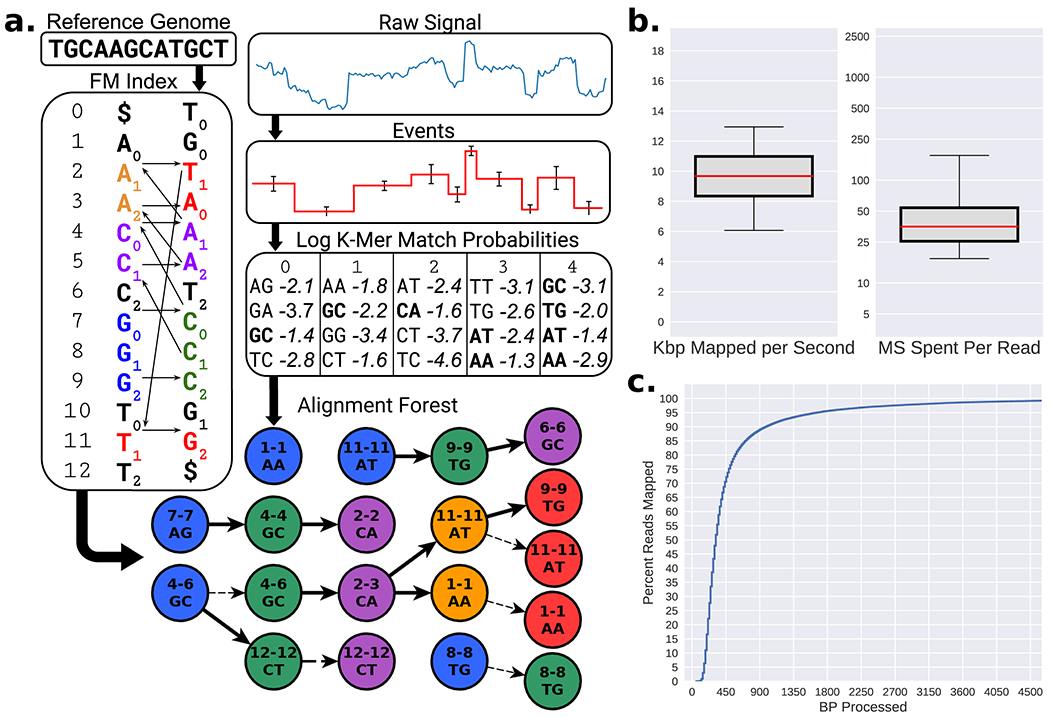
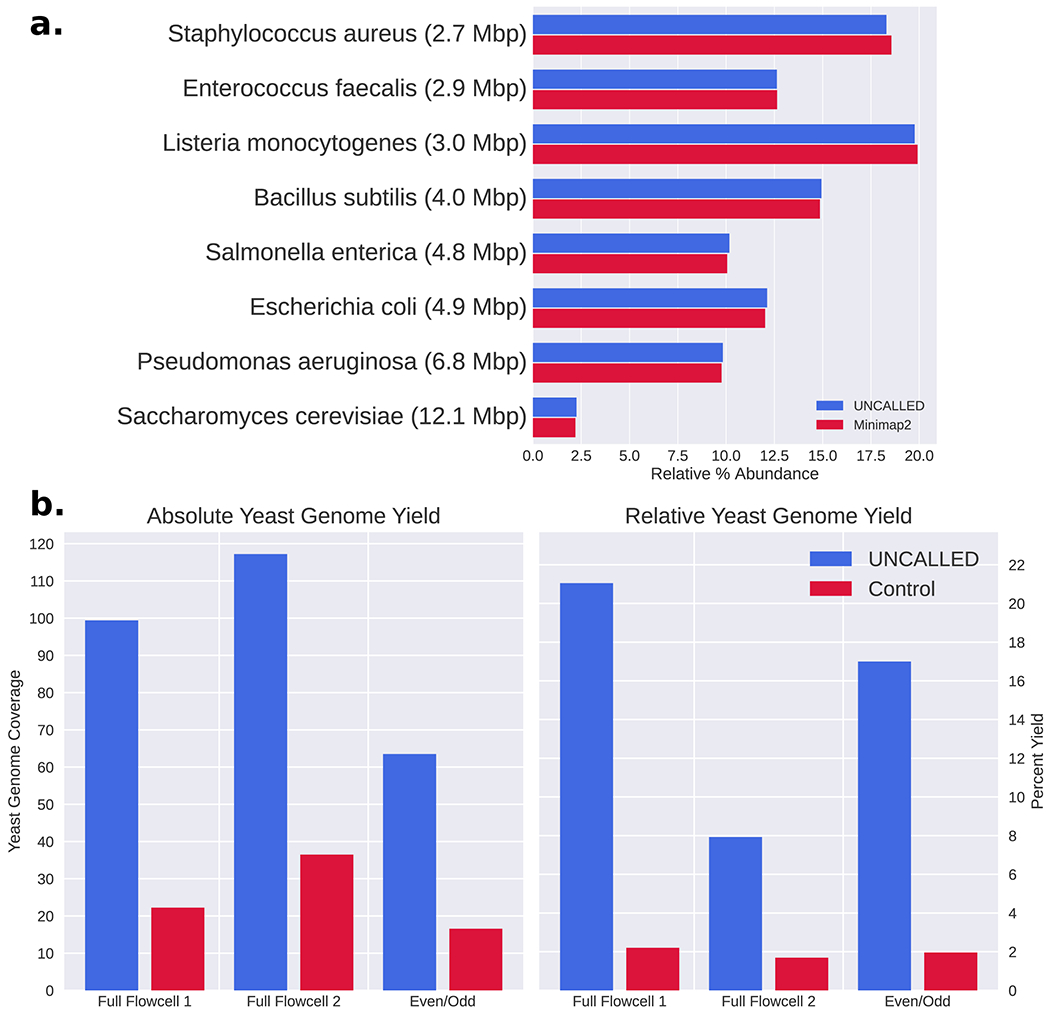
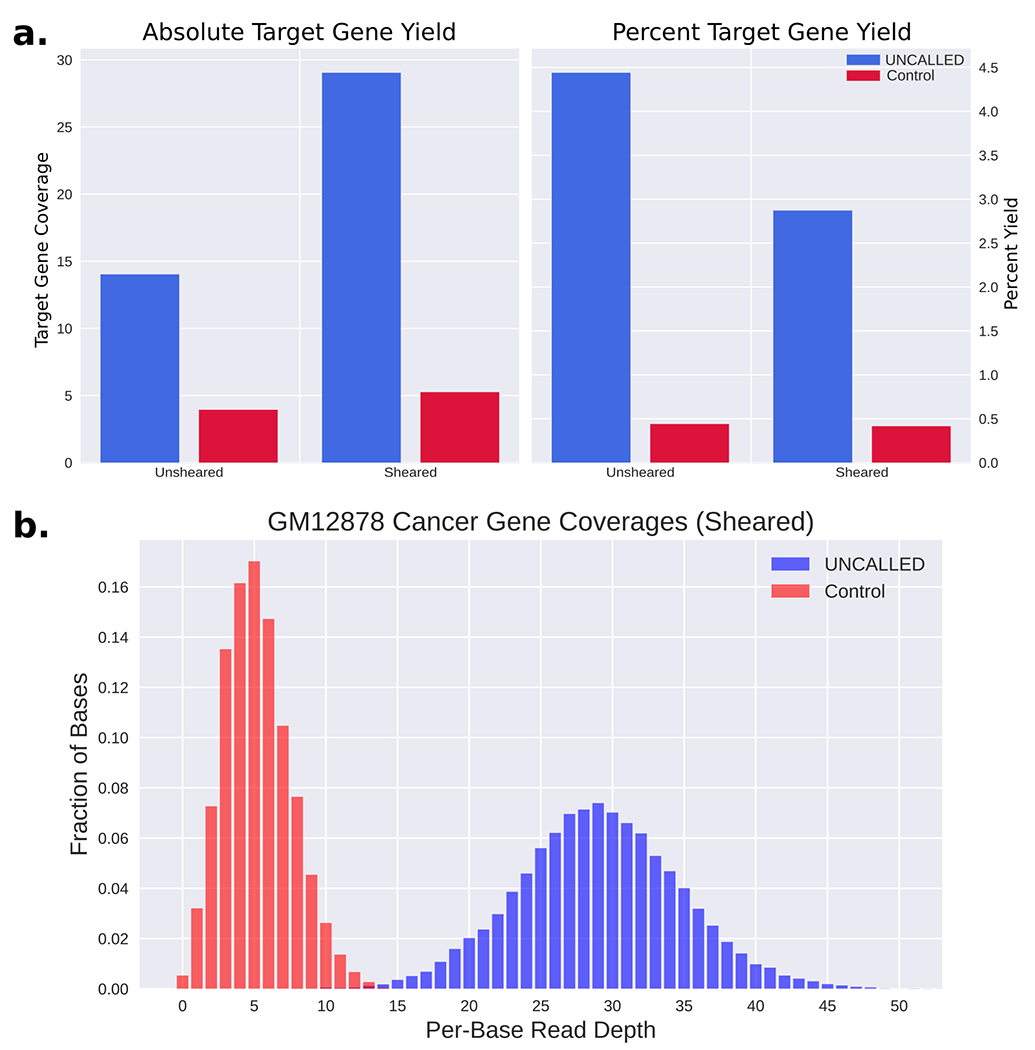
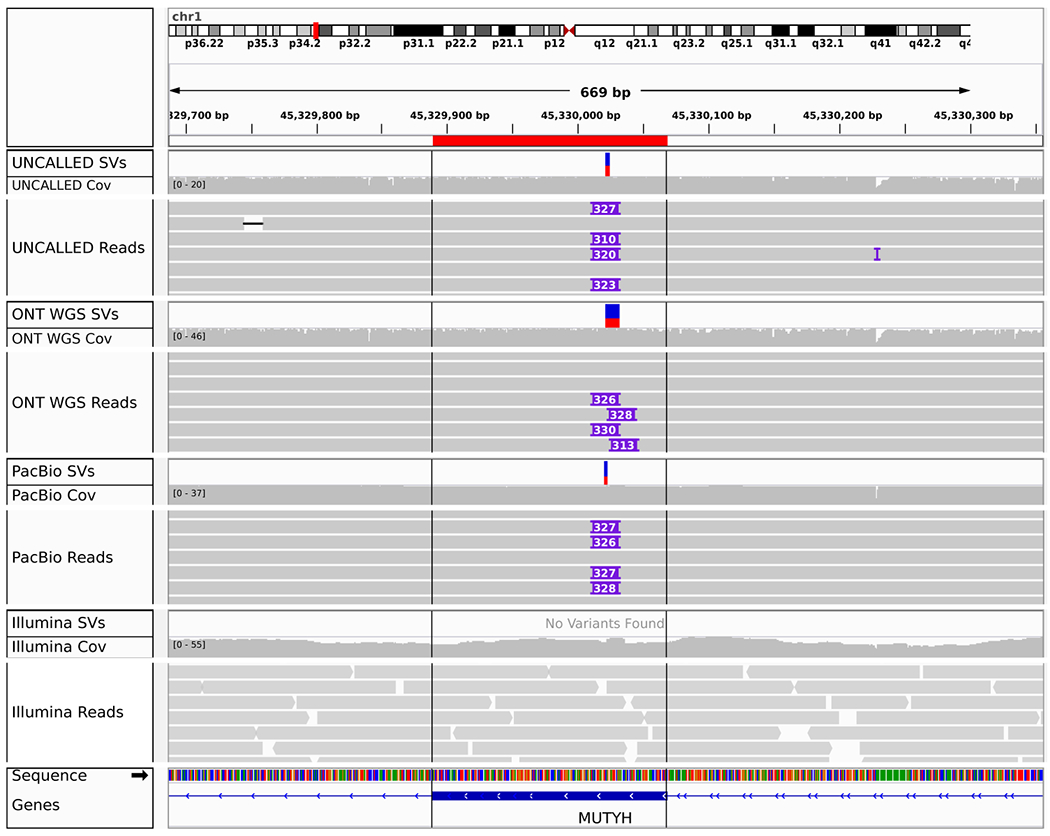
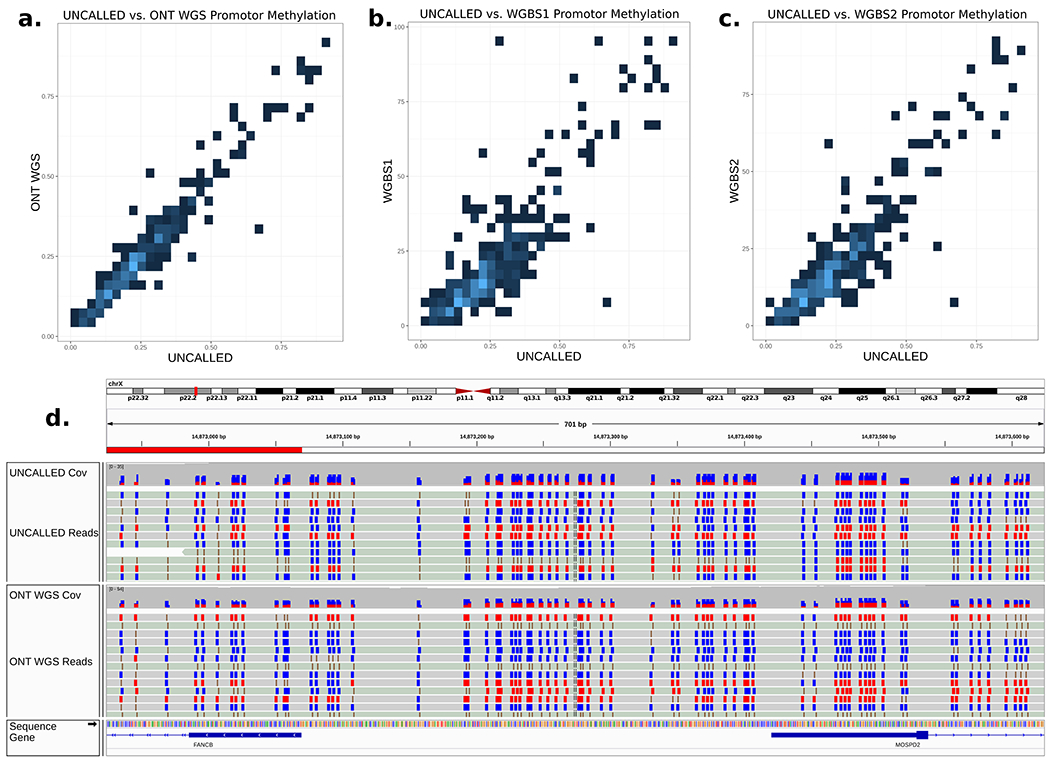
Conventional targeted sequencing methods eliminate many of the benefits of nanopore sequencing, such as the ability to accurately detect structural variants or epigenetic modifications. The ReadUntil method allows nanopore devices to selectively eject reads from pores in real time, which could enable purely computational targeted sequencing. However, this requires rapid identification of on-target reads while most mapping methods require computationally intensive basecalling. We present UNCALLED ( https://github.com/skovaka/UNCALLED ), an open source mapper that rapidly matches streaming of nanopore current signals to a reference sequence. UNCALLED probabilistically considers k-mers that could be represented by the signal and then prunes the candidates based on the reference encoded within a Ferragina-Manzini index. We used UNCALLED to deplete sequencing of known bacterial genomes within a metagenomics community, enriching the remaining species 4.46-fold. UNCALLED also enriched 148 human genes associated with hereditary cancers to 29.6× coverage using one MinION flowcell, enabling accurate detection of single-nucleotide polymorphisms, insertions and deletions, structural variants and methylation in these genes.
Figures
Comment in
-
Complex targeted sequencing in real time.Nat Rev Genet. 2021 Feb;22(2):67. doi: 10.1038/s41576-020-00324-6. Nat Rev Genet. 2021. PMID: 33349697 No abstract available.
References
References (Online Methods)
-
- Welford BP Note on a Method for Calculating Corrected Sums of Squares and Products. Technometrics 4, 419–420 (1962).
-
- Gog S & Petri M Optimized succinct data structures for massive data. Softw. Pract. Exp 44, 1287–1314 (2014).
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
