

Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements
- PMID: 33257898
- PMCID: PMC8049522
- DOI: 10.1038/s41588-020-00740-8
Improving the trans-ancestry portability of polygenic risk scores by prioritizing variants in predicted cell-type-specific regulatory elements
Abstract
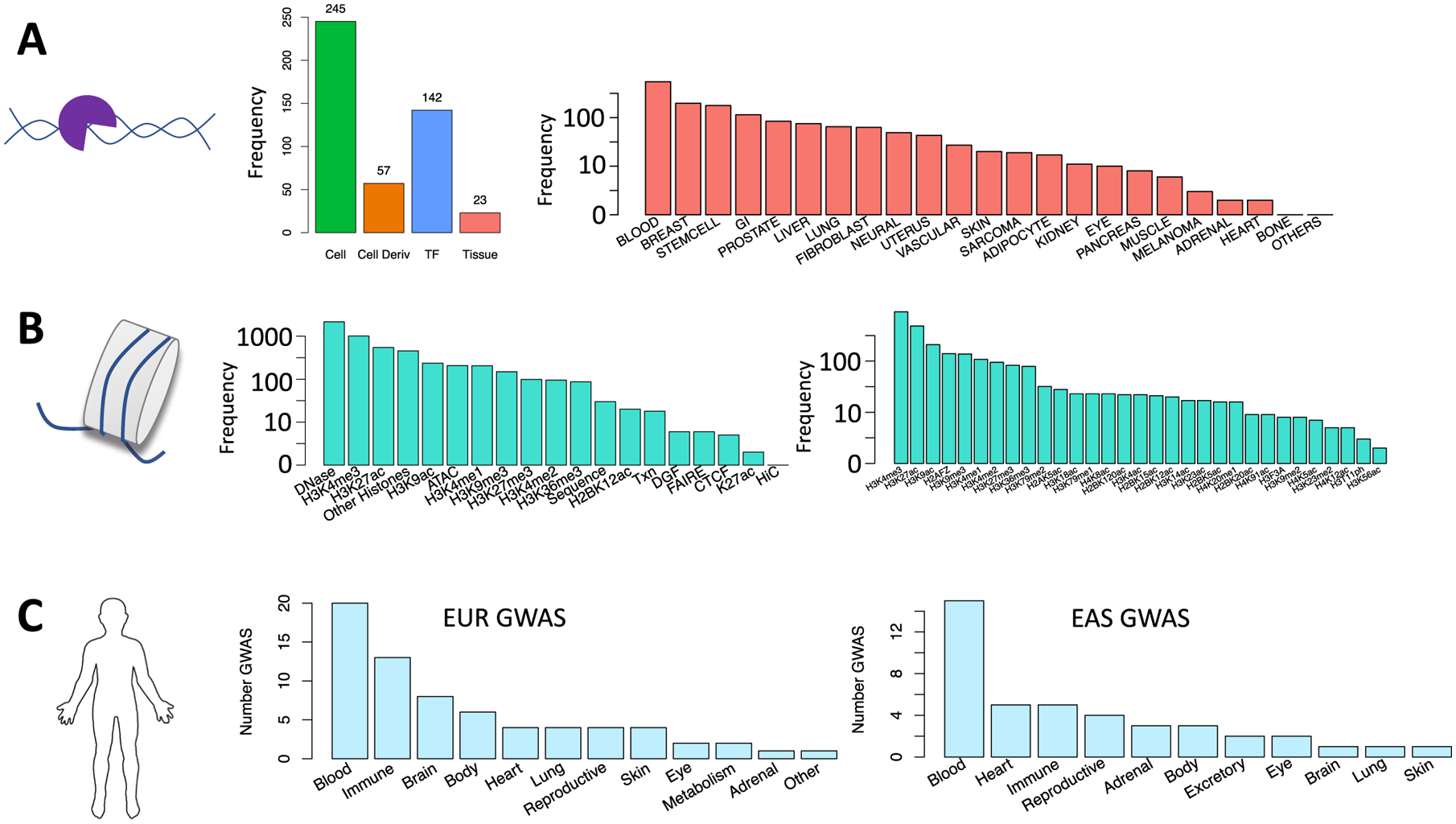
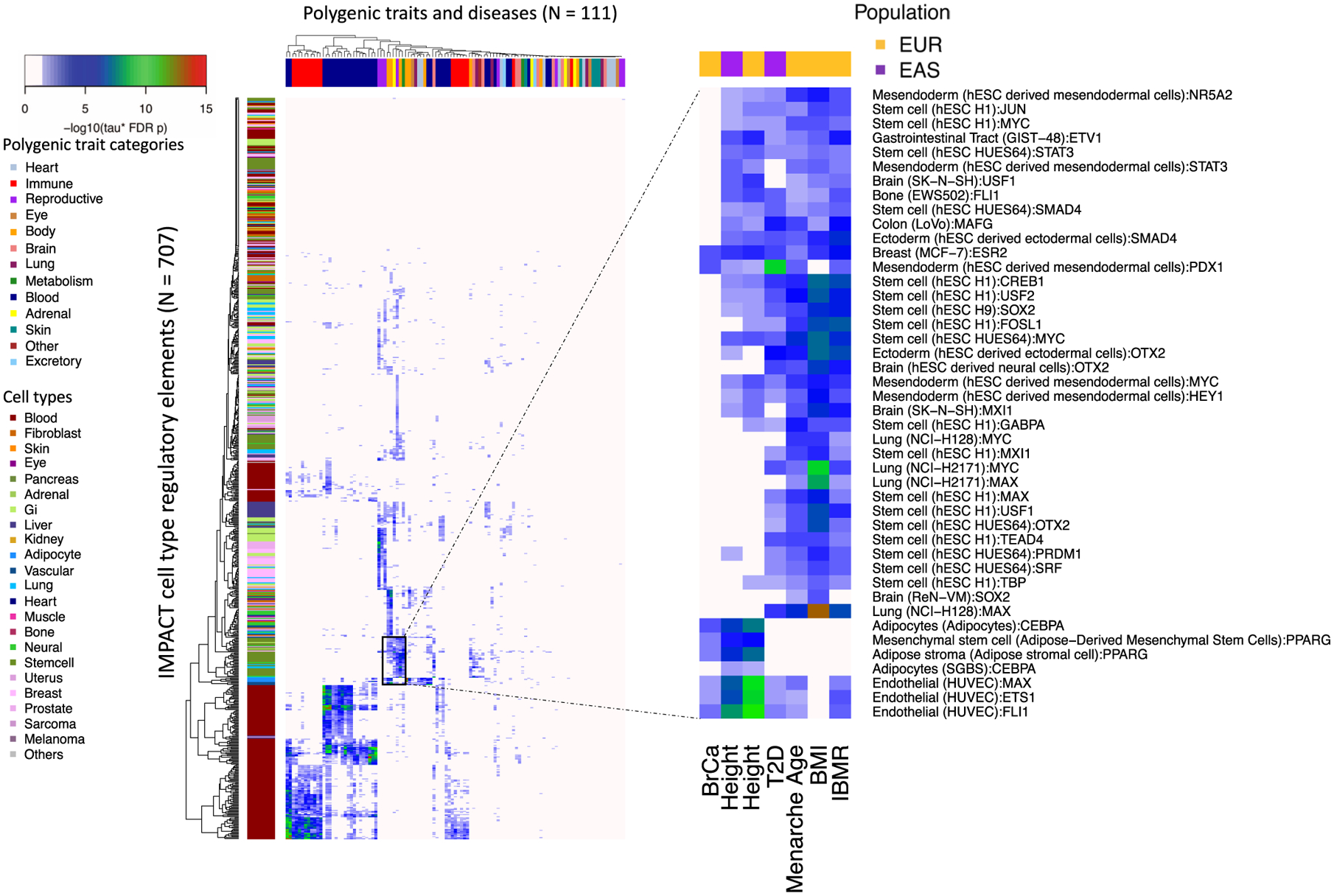
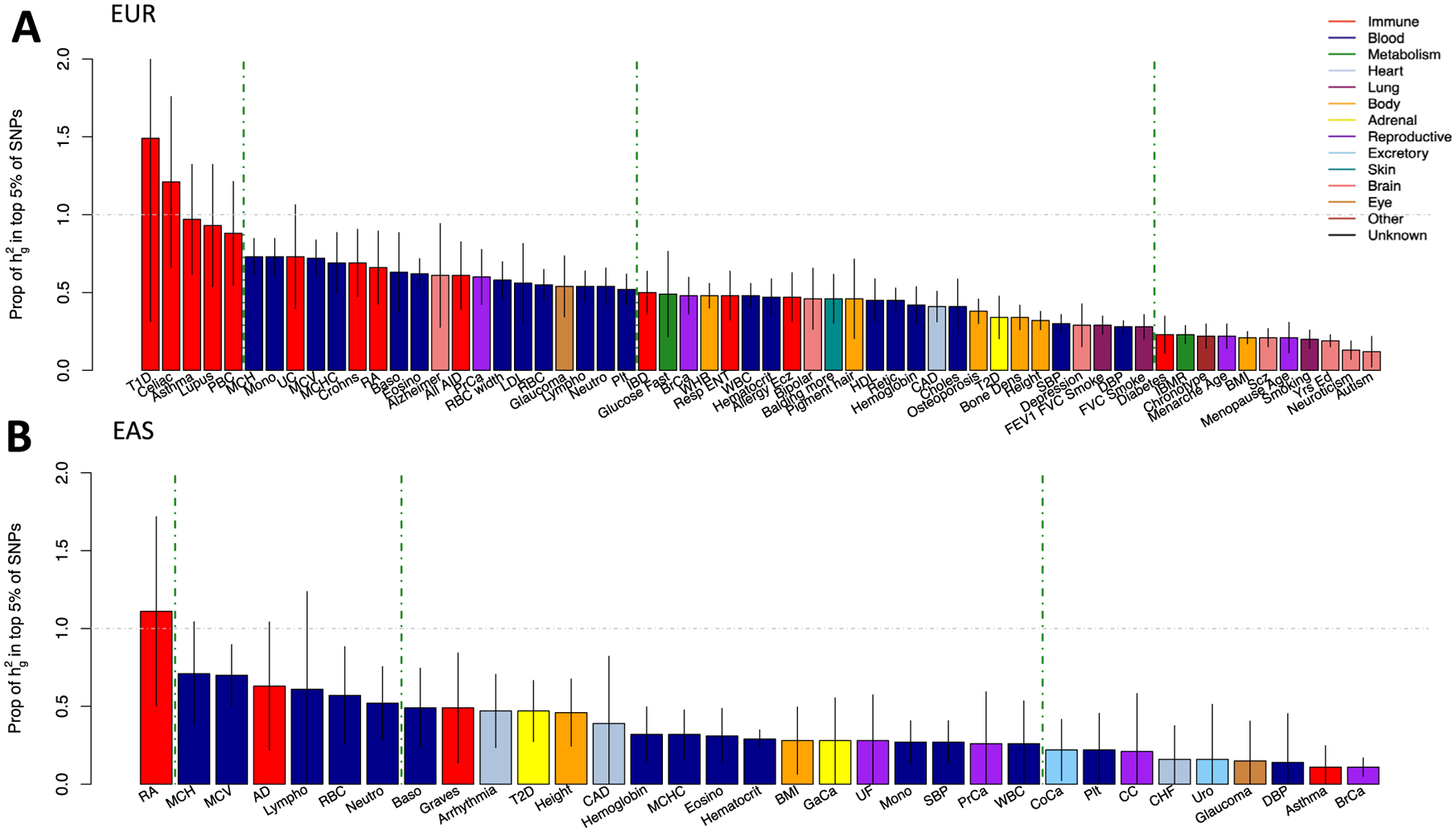
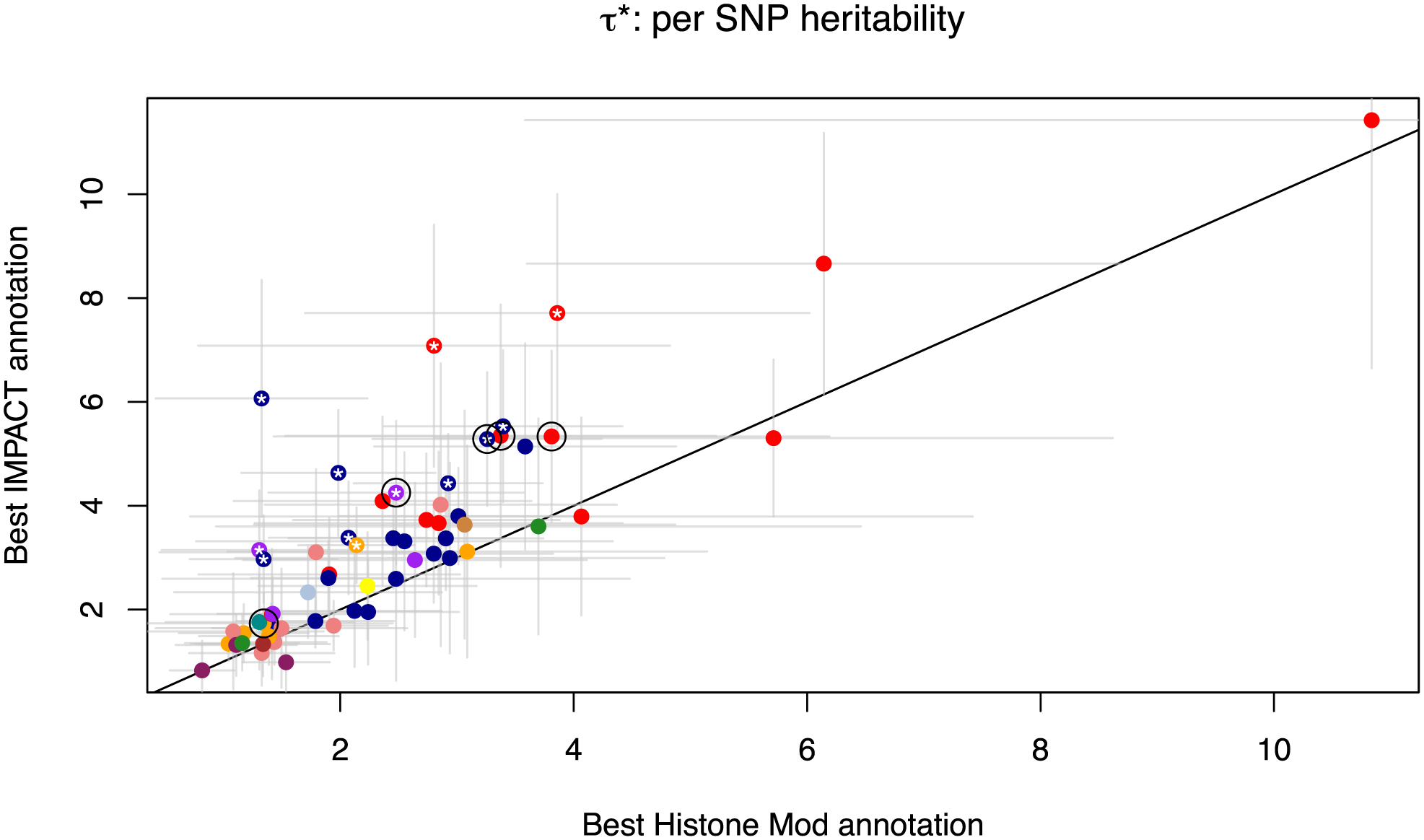
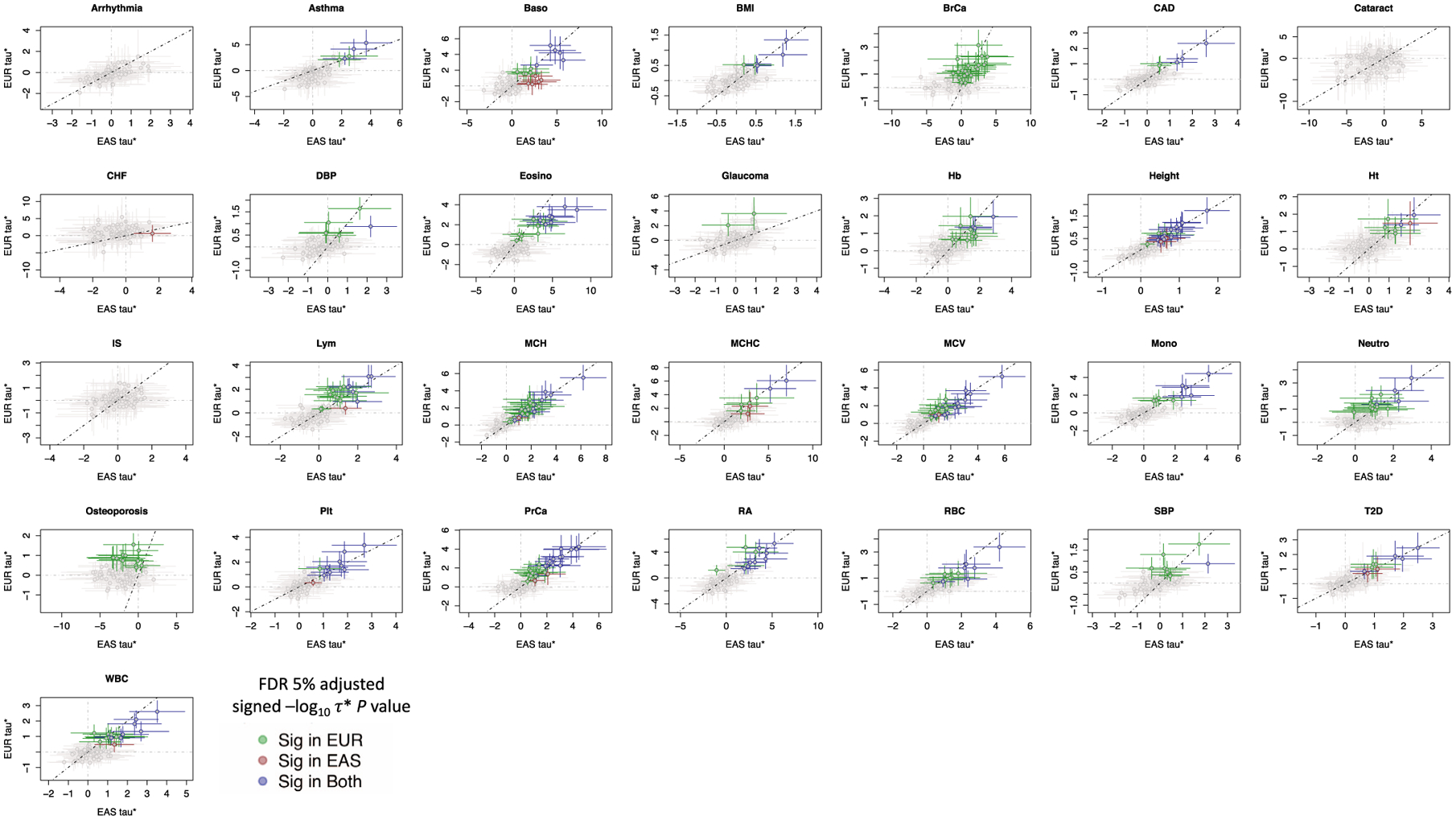
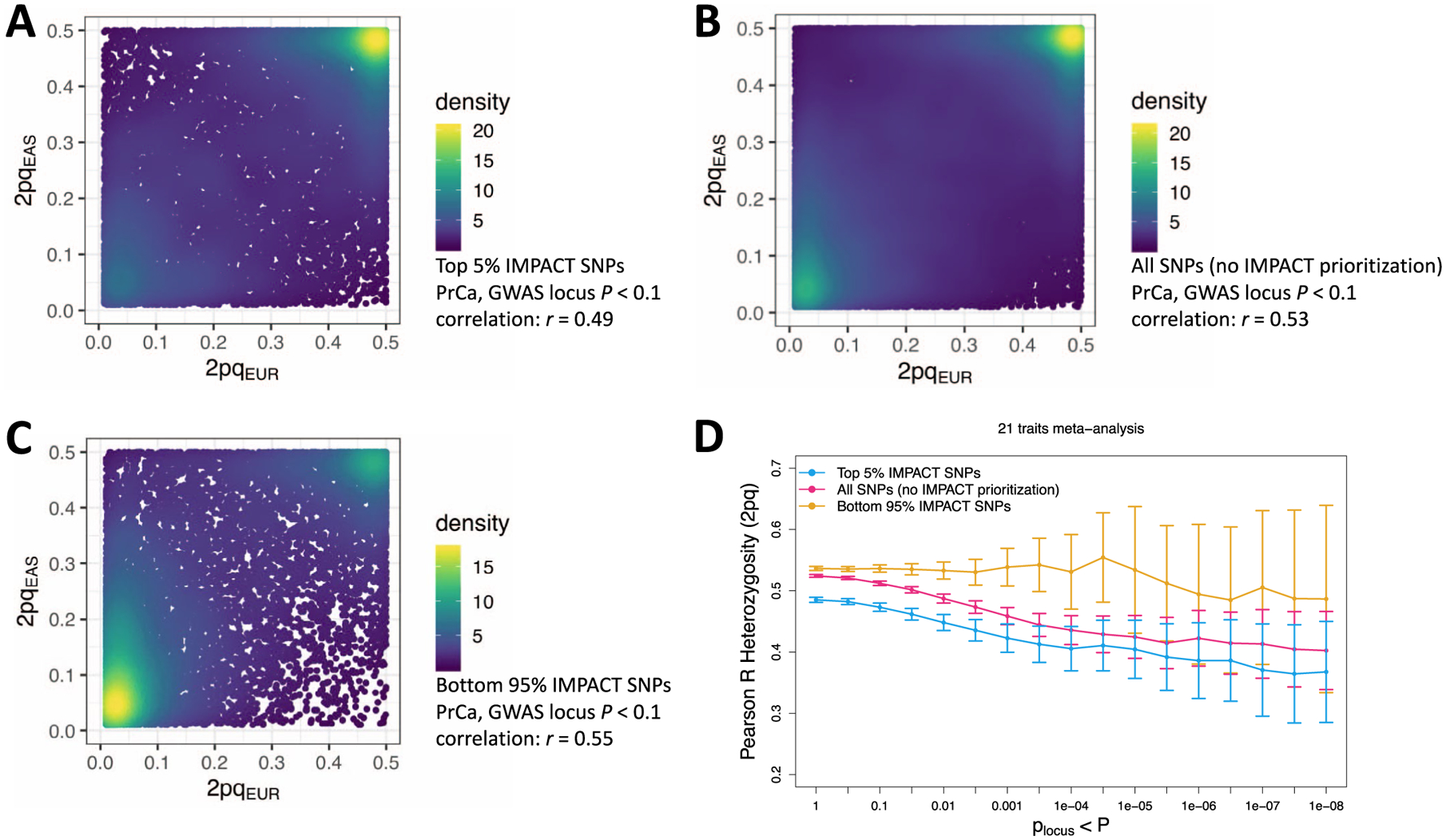
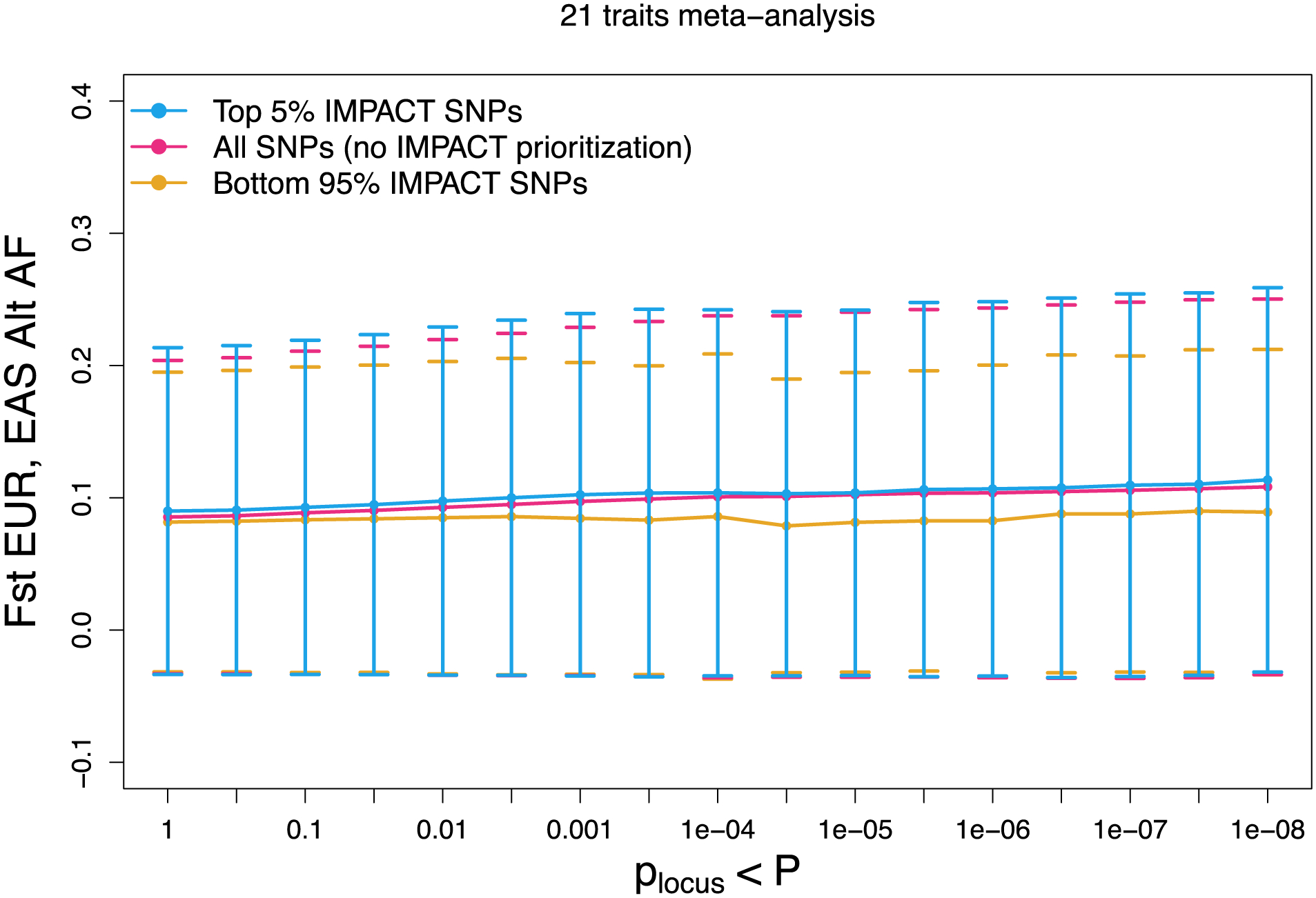
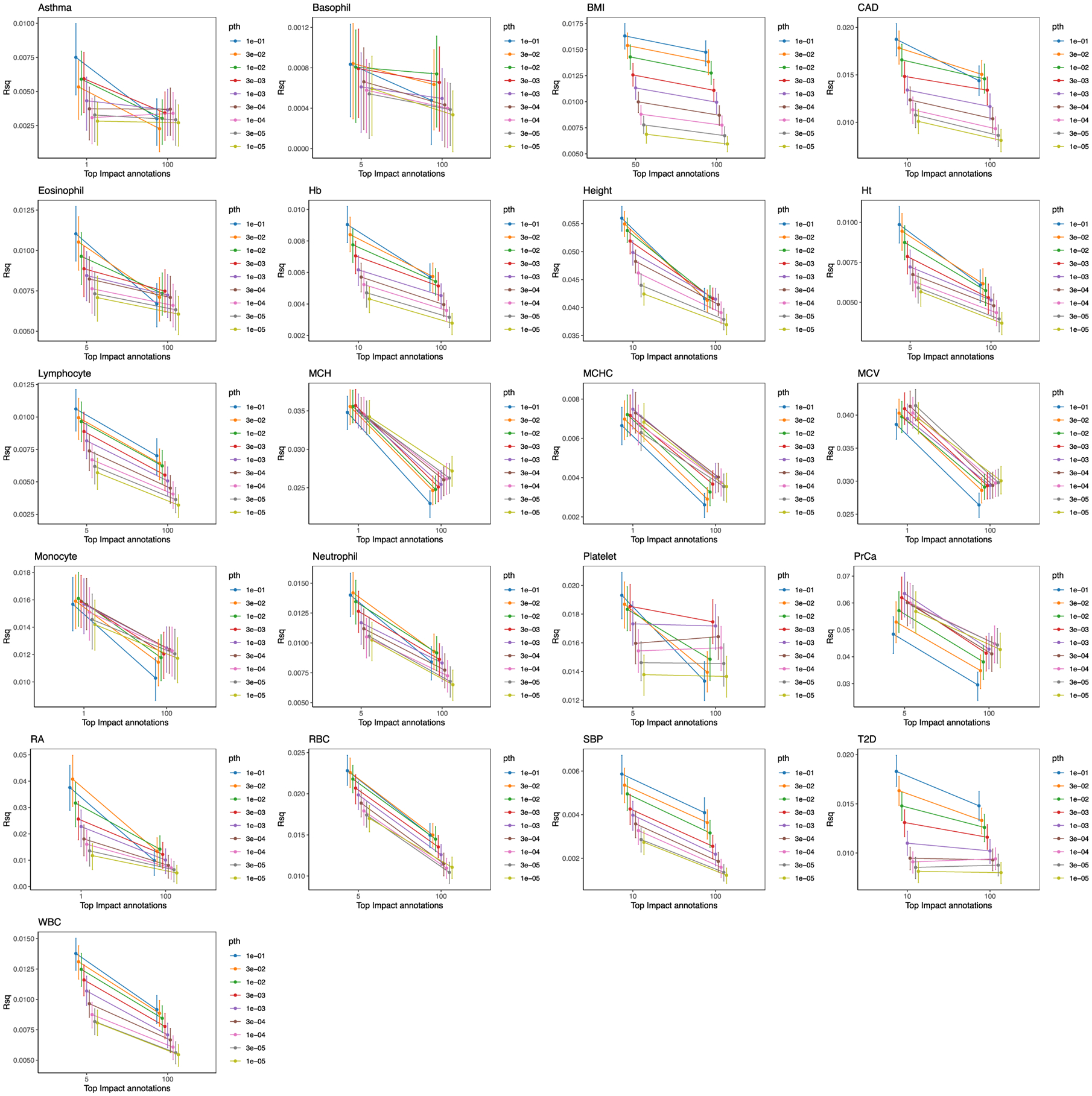
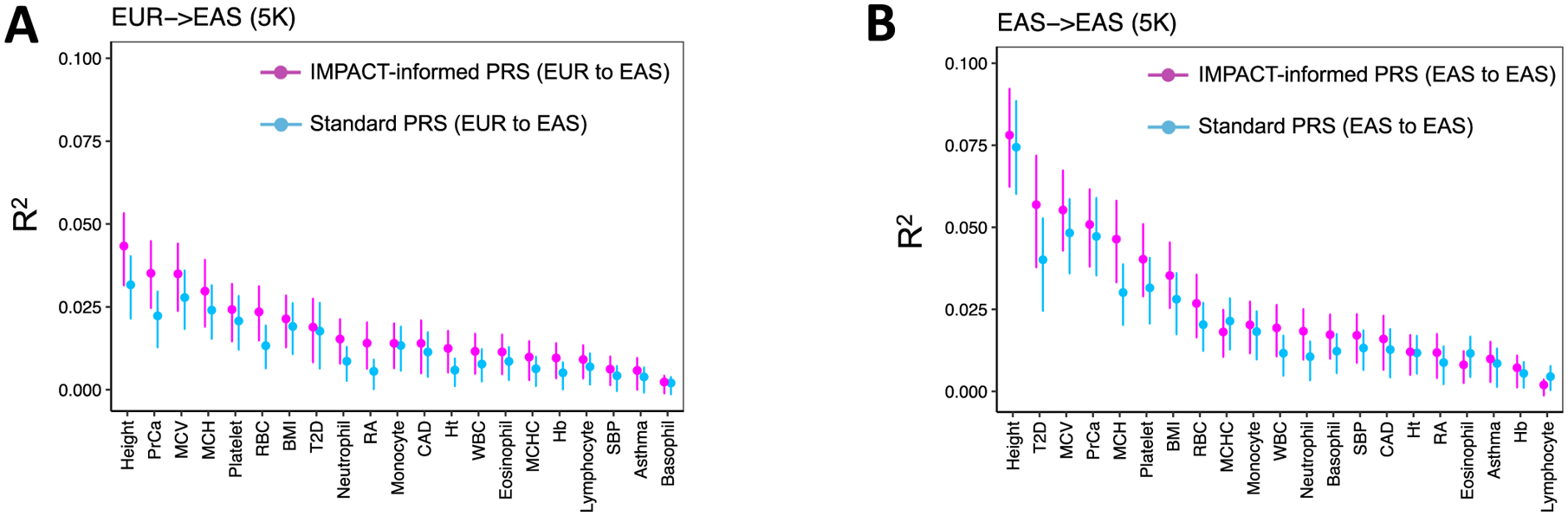
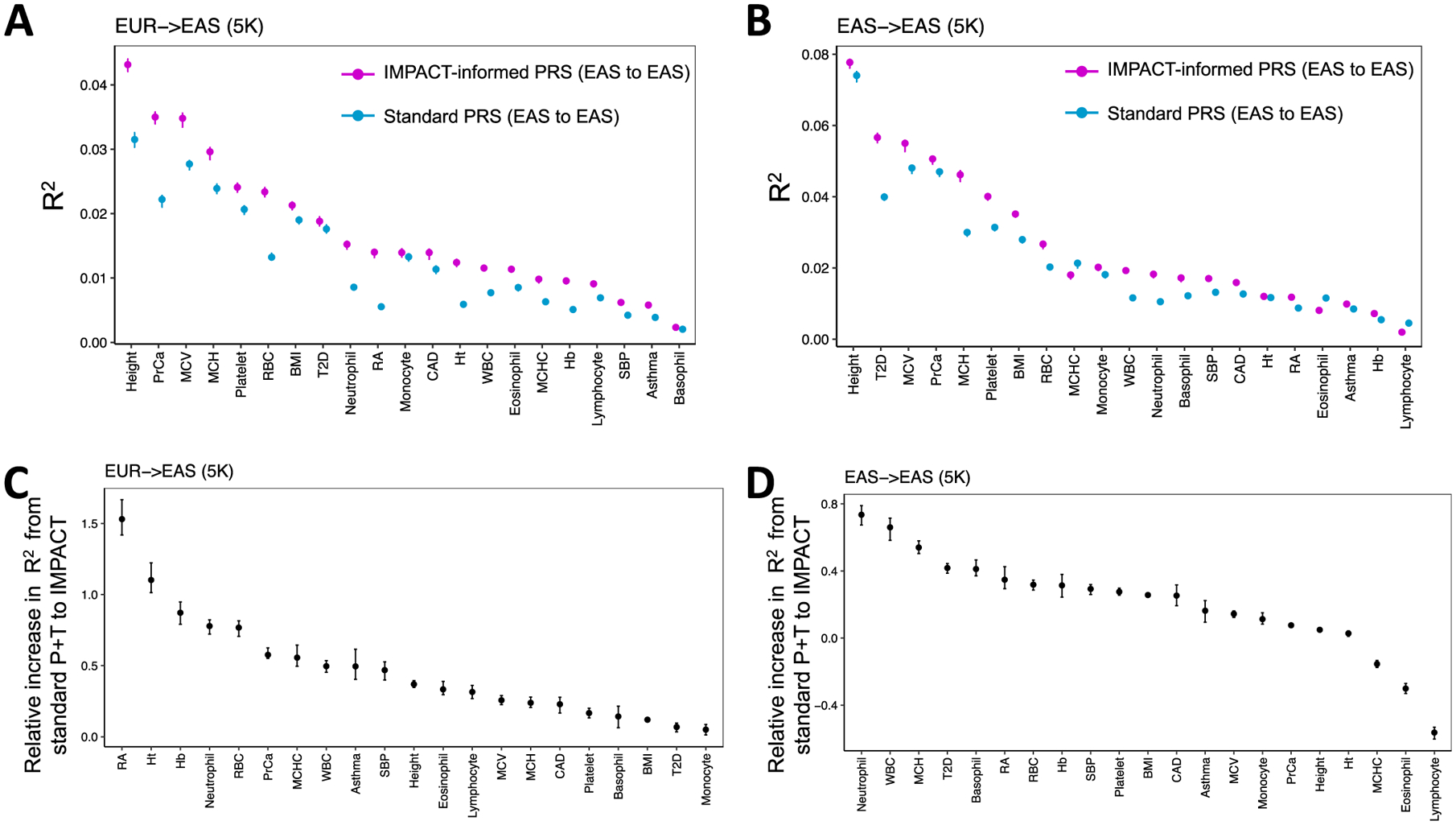
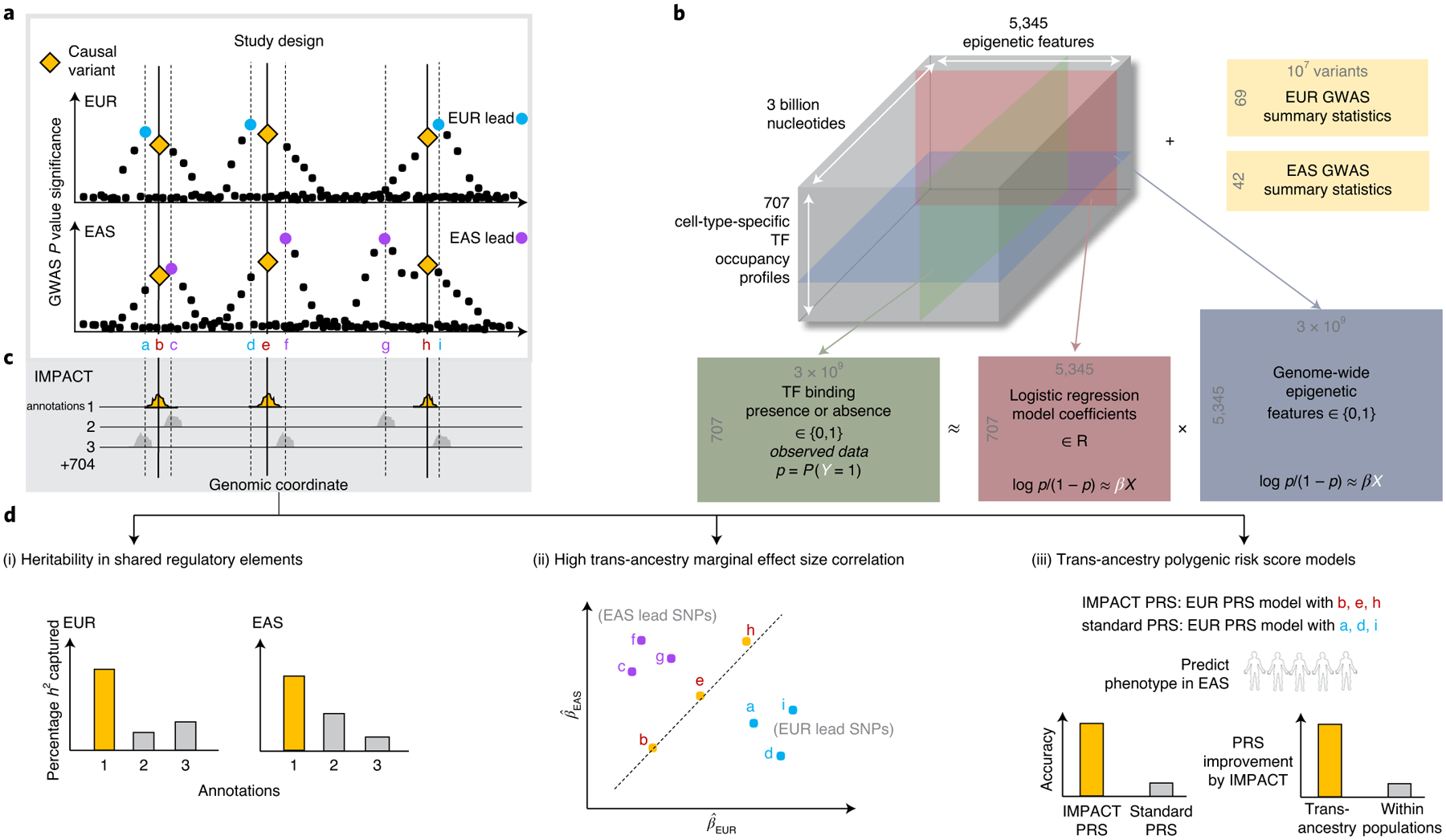
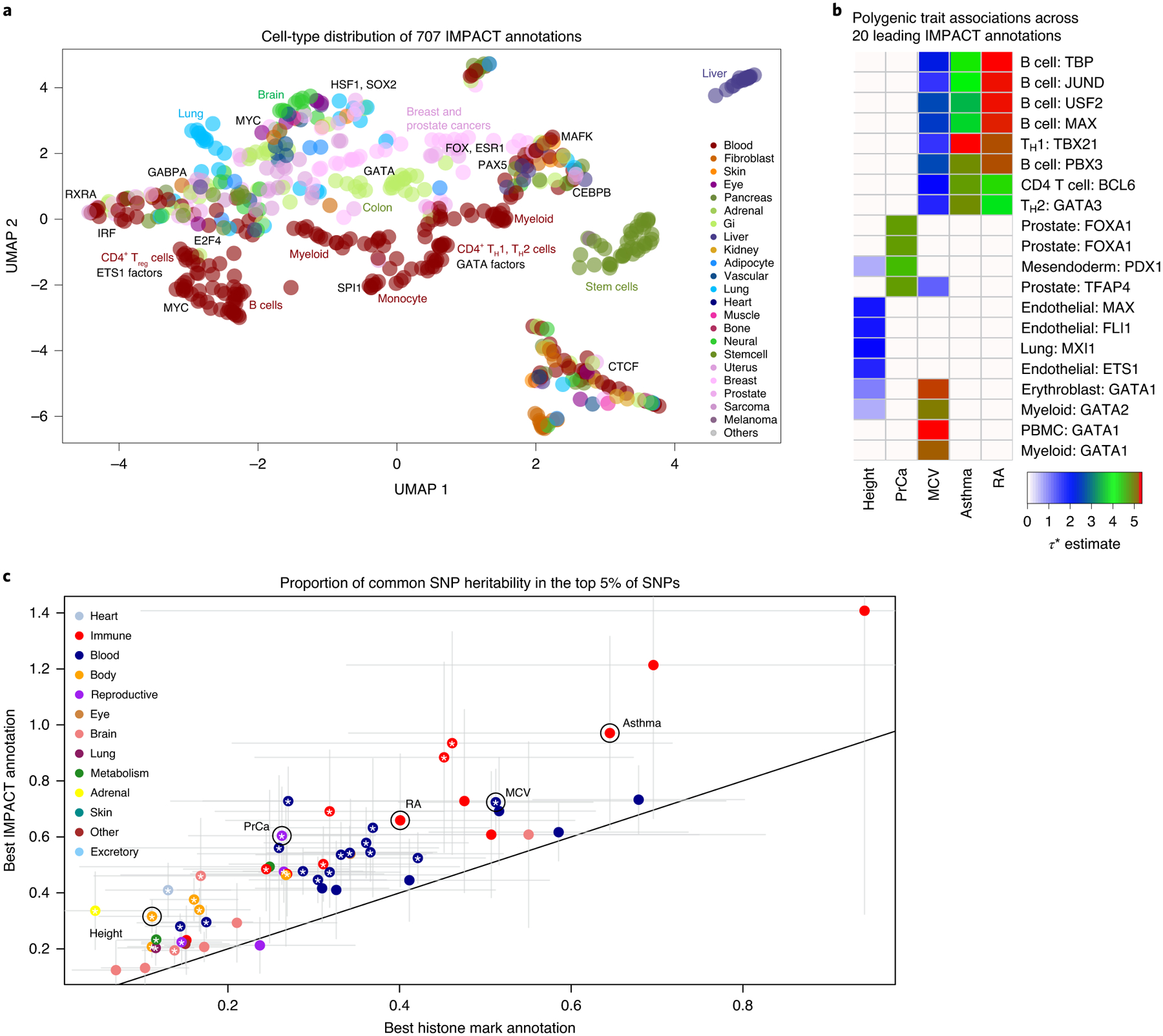
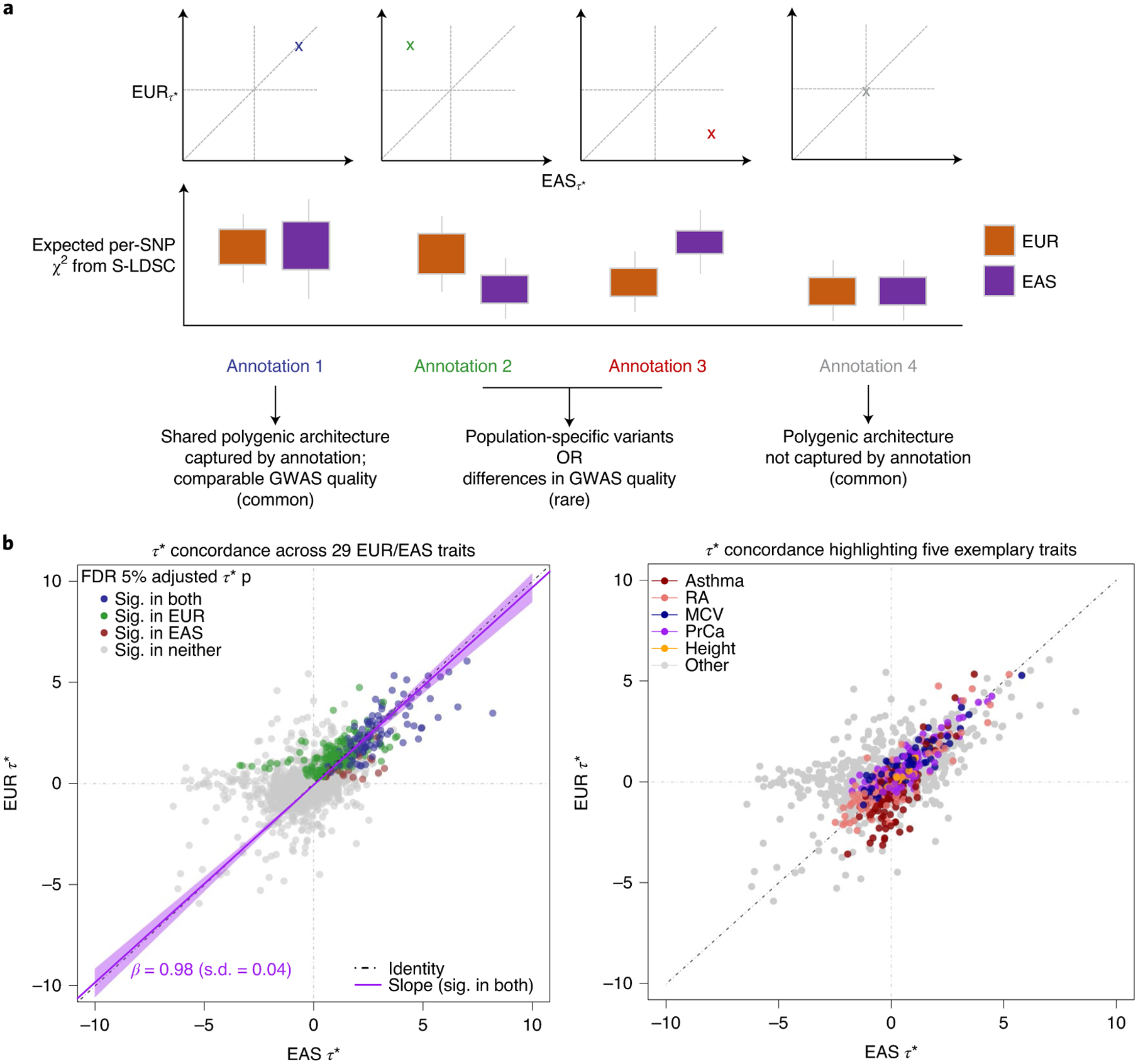
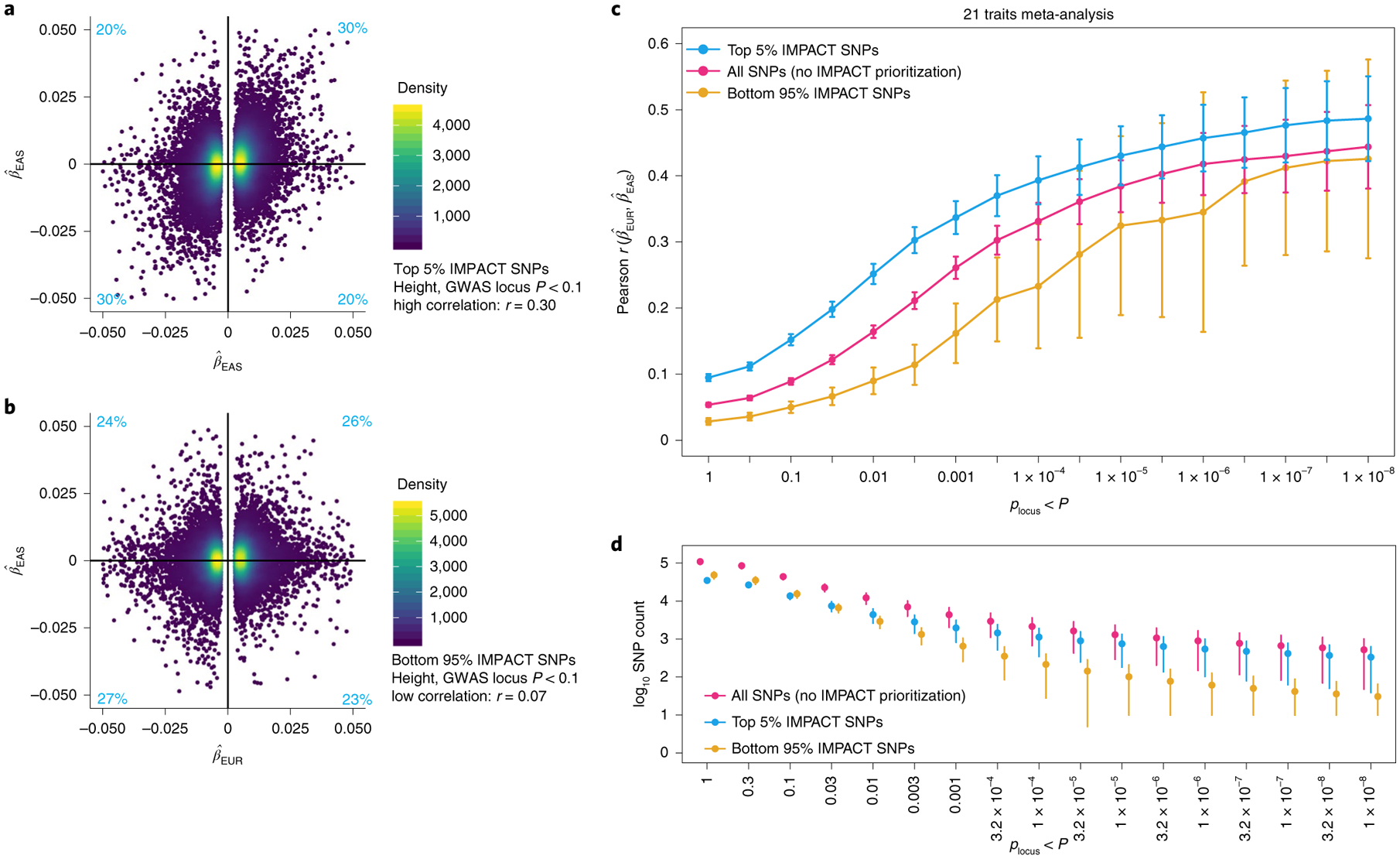
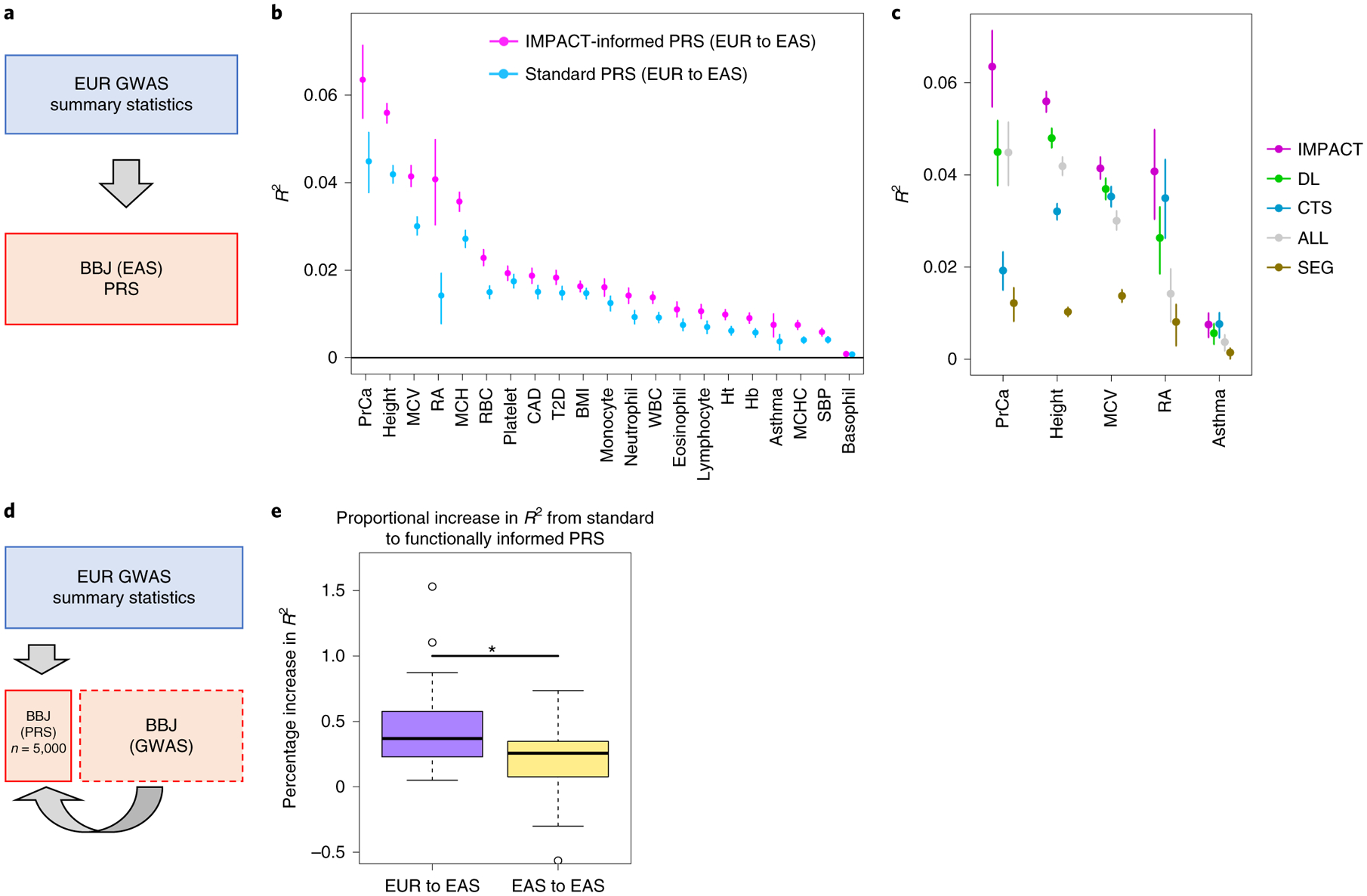
Poor trans-ancestry portability of polygenic risk scores is a consequence of Eurocentric genetic studies and limited knowledge of shared causal variants. Leveraging regulatory annotations may improve portability by prioritizing functional over tagging variants. We constructed a resource of 707 cell-type-specific IMPACT regulatory annotations by aggregating 5,345 epigenetic datasets to predict binding patterns of 142 transcription factors across 245 cell types. We then partitioned the common SNP heritability of 111 genome-wide association study summary statistics of European (average n ≈ 189,000) and East Asian (average n ≈ 157,000) origin. IMPACT annotations captured consistent SNP heritability between populations, suggesting prioritization of shared functional variants. Variant prioritization using IMPACT resulted in increased trans-ancestry portability of polygenic risk scores from Europeans to East Asians across all 21 phenotypes analyzed (49.9% mean relative increase in R2). Our study identifies a crucial role for functional annotations such as IMPACT to improve the trans-ancestry portability of genetic data.
Conflict of interest statement
Competing interests
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
