

Cellular and mitochondrial mechanisms of atrial fibrillation
- PMID: 33258071
- PMCID: PMC7704501
- DOI: 10.1007/s00395-020-00827-7
Cellular and mitochondrial mechanisms of atrial fibrillation
Abstract
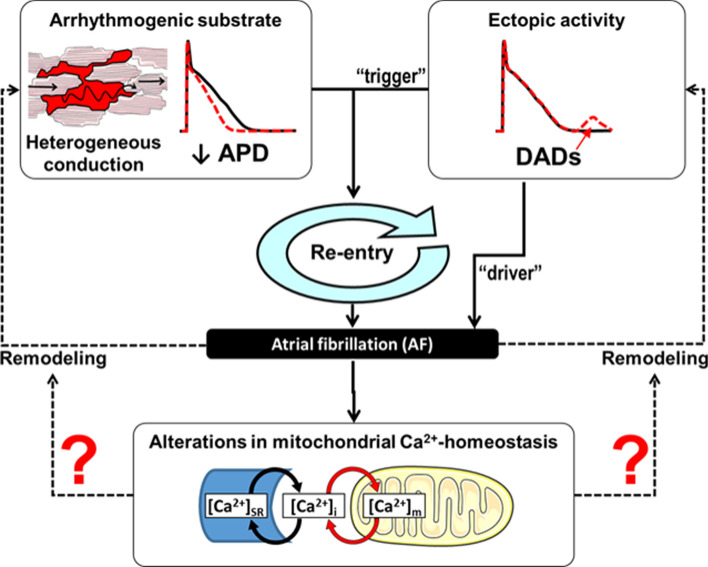
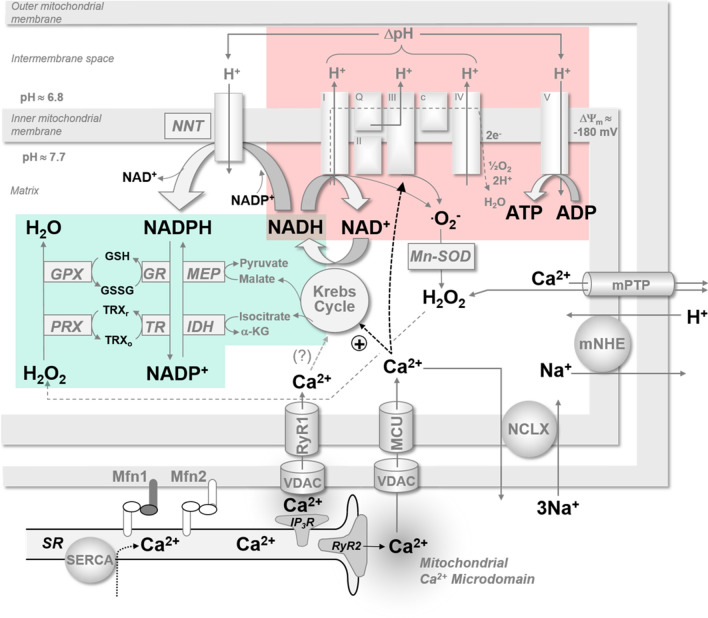
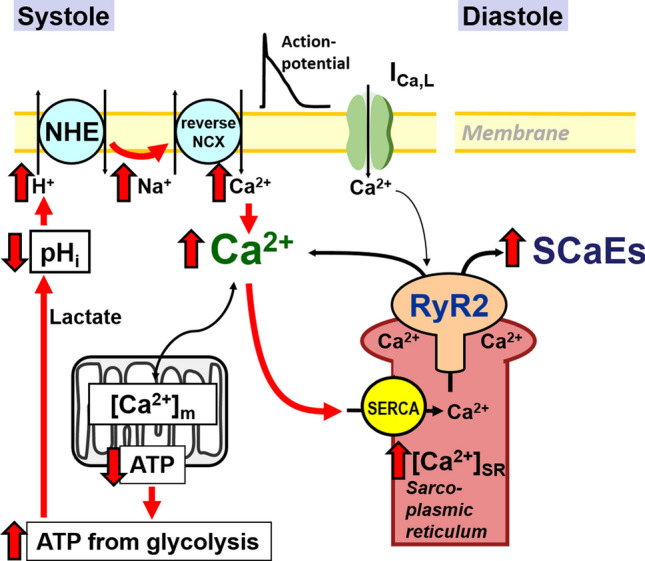
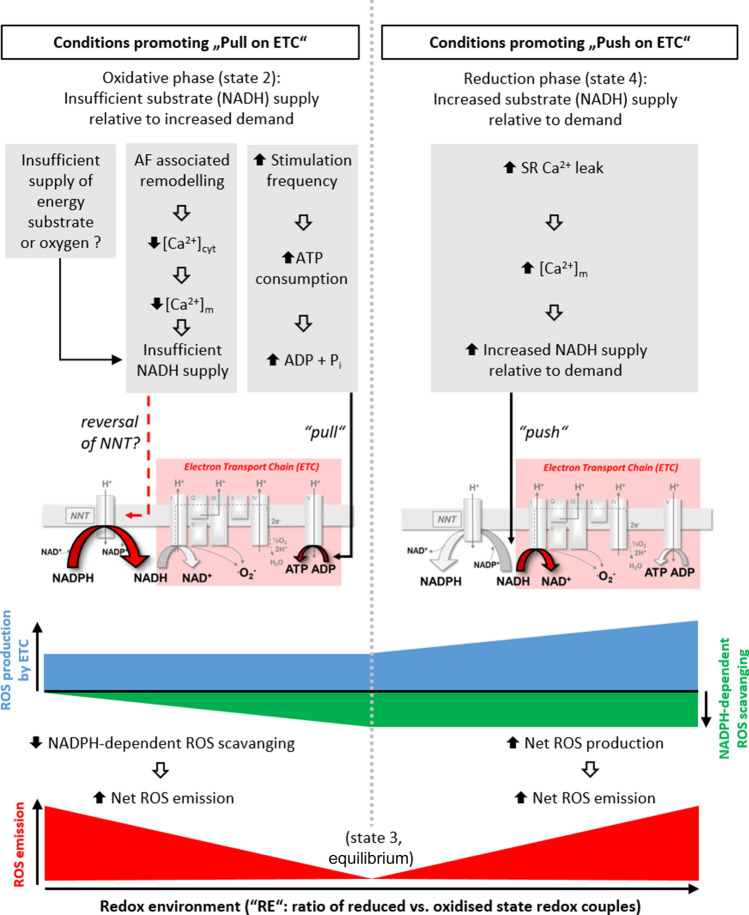
The molecular mechanisms underlying atrial fibrillation (AF), the most common form of arrhythmia, are poorly understood and therefore target-specific treatment options remain an unmet clinical need. Excitation-contraction coupling in cardiac myocytes requires high amounts of adenosine triphosphate (ATP), which is replenished by oxidative phosphorylation in mitochondria. Calcium (Ca2+) is a key regulator of mitochondrial function by stimulating the Krebs cycle, which produces nicotinamide adenine dinucleotide for ATP production at the electron transport chain and nicotinamide adenine dinucleotide phosphate for the elimination of reactive oxygen species (ROS). While it is now well established that mitochondrial dysfunction plays an important role in the pathophysiology of heart failure, this has been less investigated in atrial myocytes in AF. Considering the high prevalence of AF, investigating the role of mitochondria in this disease may guide the path towards new therapeutic targets. In this review, we discuss the importance of mitochondrial Ca2+ handling in regulating ATP production and mitochondrial ROS emission and how alterations, particularly in these aspects of mitochondrial activity, may play a role in AF. In addition to describing research advances, we highlight areas in which further studies are required to elucidate the role of mitochondria in AF.
Keywords: Atrial cardiomyopathy; Atrial fibrillation; Calcium; Electrophysiology; Mitochondria; Oxidative stress.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
-
- Ad N, Schneider A, Khaliulin I, Borman JB, Schwalb H. Impaired mitochondrial response to simulated ischemic injury as a predictor of the development of atrial fibrillation after cardiac surgery: in vitro study in human myocardium. J Thorac Cardiovasc Surg. 2005;129:41–45. doi: 10.1016/j.jtcvs.2004.03.058. - DOI - PubMed
-
- Anderson EJ, Efird JT, Davies SW, O’Neal WT, Darden TM, Thayne KA, Katunga LA, Kindell LC, Ferguson TB, Anderson CA, Chitwood WR, Koutlas TC, Williams JM, Rodriguez E, Kypson AP. Monoamine oxidase is a major determinant of redox balance in human atrial myocardium and is associated with postoperative atrial fibrillation. J Am Heart Assoc. 2014;3:e000713. doi: 10.1161/JAHA.113.000713. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- VO 1568/3-1/Deutsche Forschungsgemeinschaft/International
- IRTG1816/Deutsche Forschungsgemeinschaft/International
- SFB1002/Deutsche Forschungsgemeinschaft/International
- EXC 2067/1- 390729940/Deutsche Forschungsgemeinschaft/International
- Ma 2528/7-1/Deutsche Forschungsgemeinschaft/International
- SFB 894/Deutsche Forschungsgemeinschaft/International
- TRR-219/Deutsche Forschungsgemeinschaft/International
- EKFS 2016_A20/Else Kröner-Fresenius-Stiftung/International
- DZHK GOE MD3/Deutsches Zentrum für Herz-Kreislaufforschung/International
- SE181/Deutsches Zentrum für Herz-Kreislaufforschung/International
- BMBF/Bundesministerium für Bildung und Forschung/International
- 01EO1504/Bundesministerium für Bildung und Forschung/International
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
