

Reduced reticulum-mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy
- PMID: 33258101
- PMCID: PMC7704523
- DOI: 10.1007/s00395-020-00835-7
Reduced reticulum-mitochondria Ca2+ transfer is an early and reversible trigger of mitochondrial dysfunctions in diabetic cardiomyopathy
Abstract
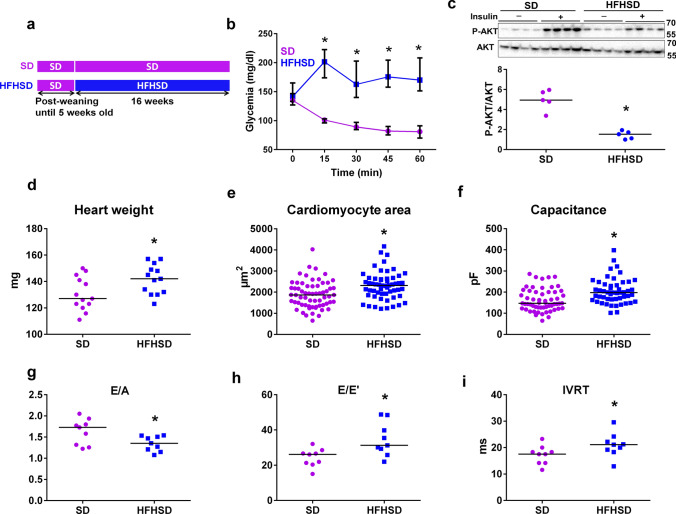
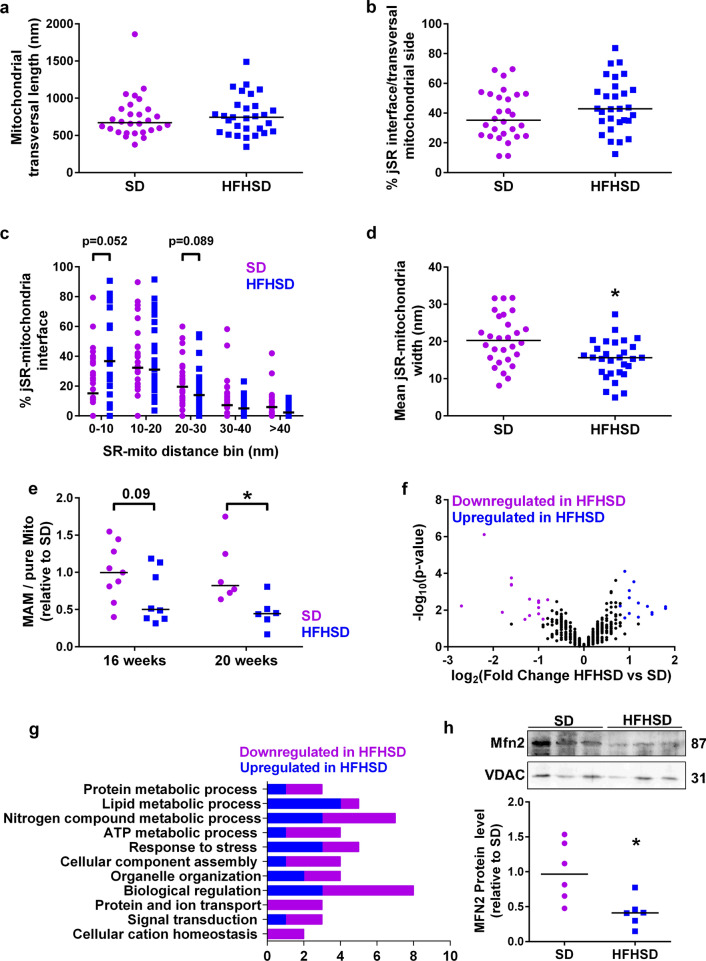
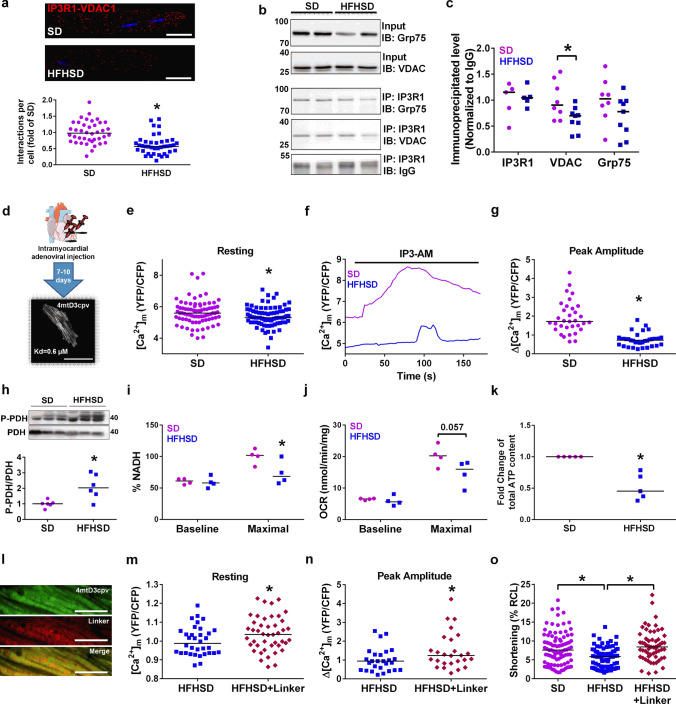
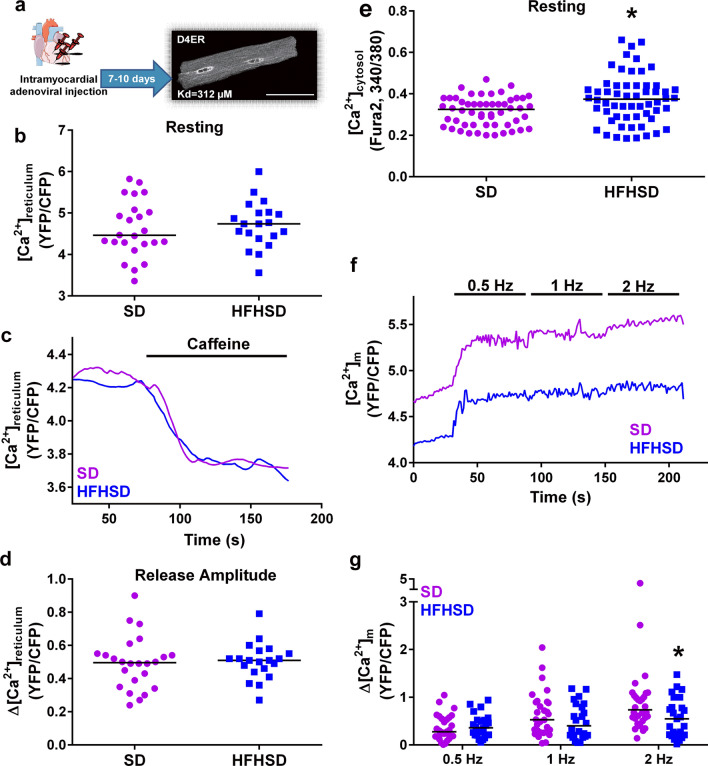
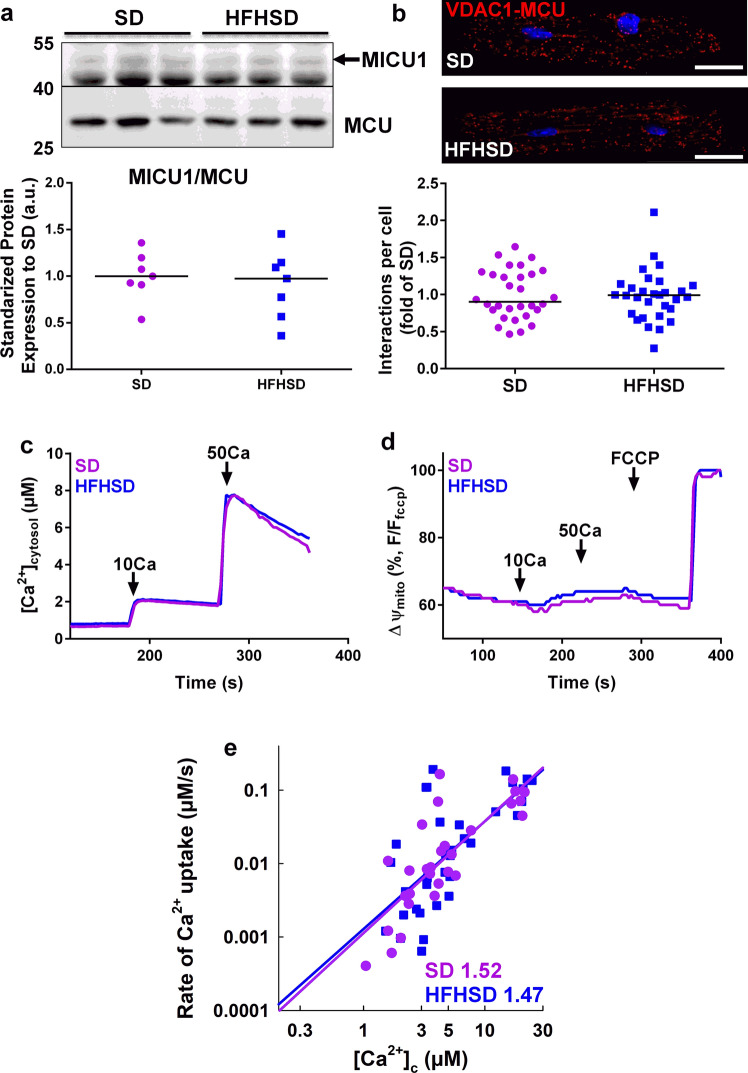
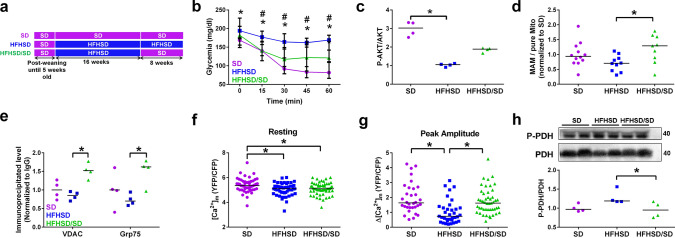
Type 2 diabetic cardiomyopathy features Ca2+ signaling abnormalities, notably an altered mitochondrial Ca2+ handling. We here aimed to study if it might be due to a dysregulation of either the whole Ca2+ homeostasis, the reticulum-mitochondrial Ca2+ coupling, and/or the mitochondrial Ca2+ entry through the uniporter. Following a 16-week high-fat high-sucrose diet (HFHSD), mice developed cardiac insulin resistance, fibrosis, hypertrophy, lipid accumulation, and diastolic dysfunction when compared to standard diet. Ultrastructural and proteomic analyses of cardiac reticulum-mitochondria interface revealed tighter interactions not compatible with Ca2+ transport in HFHSD cardiomyocytes. Intramyocardial adenoviral injections of Ca2+ sensors were performed to measure Ca2+ fluxes in freshly isolated adult cardiomyocytes and to analyze the direct effects of in vivo type 2 diabetes on cardiomyocyte function. HFHSD resulted in a decreased IP3R-VDAC interaction and a reduced IP3-stimulated Ca2+ transfer to mitochondria, with no changes in reticular Ca2+ level, cytosolic Ca2+ transients, and mitochondrial Ca2+ uniporter function. Disruption of organelle Ca2+ exchange was associated with decreased mitochondrial bioenergetics and reduced cell contraction, which was rescued by an adenovirus-mediated expression of a reticulum-mitochondria linker. An 8-week diet reversal was able to restore cardiac insulin signaling, Ca2+ transfer, and cardiac function in HFHSD mice. Therefore, our study demonstrates that the reticulum-mitochondria Ca2+ miscoupling may play an early and reversible role in the development of diabetic cardiomyopathy by disrupting primarily the mitochondrial bioenergetics. A diet reversal, by counteracting the MAM-induced mitochondrial Ca2+ dysfunction, might contribute to restore normal cardiac function and prevent the exacerbation of diabetic cardiomyopathy.
Keywords: Ca2+ flux; Diabetic cardiomyopathy; Metabolic syndrome disease; Mitochondria-associated membranes MAM; Protein database; Proteomic analysis of cardiac MAM proteome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Bartok A, Weaver D, Golenar T, Nichtova Z, Katona M, Bansaghi S, Alzayady KJ, Thomas VK, Ando H, Mikoshiba K, Joseph SK, Yule DI, Csordas G, Hajnoczky G. IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat Commun. 2019;10:3726. doi: 10.1038/s41467-019-11646-3. - DOI - PMC - PubMed
-
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
