

Obesity, kidney dysfunction, and inflammation: interactions in hypertension
- PMID: 33258945
- PMCID: PMC8262632
- DOI: 10.1093/cvr/cvaa336
Obesity, kidney dysfunction, and inflammation: interactions in hypertension
Abstract
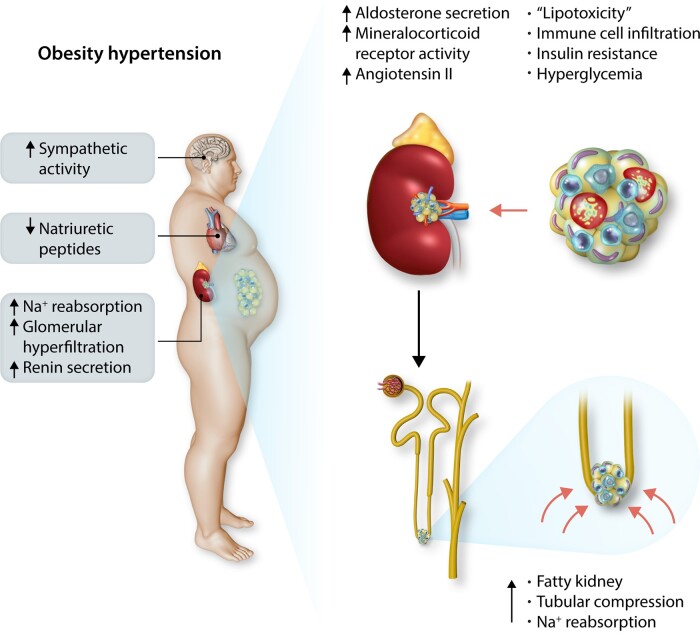
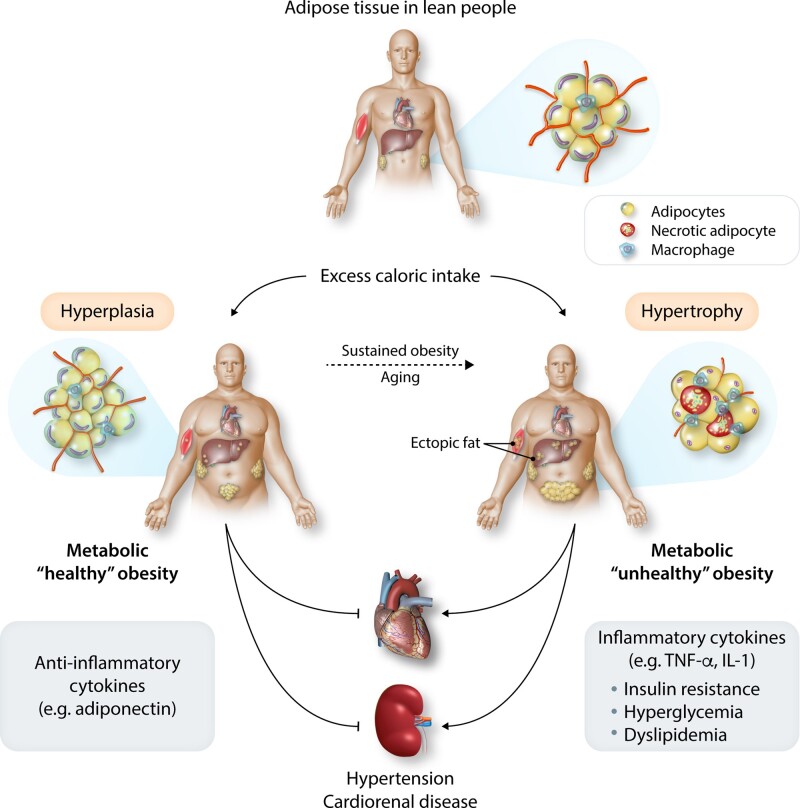
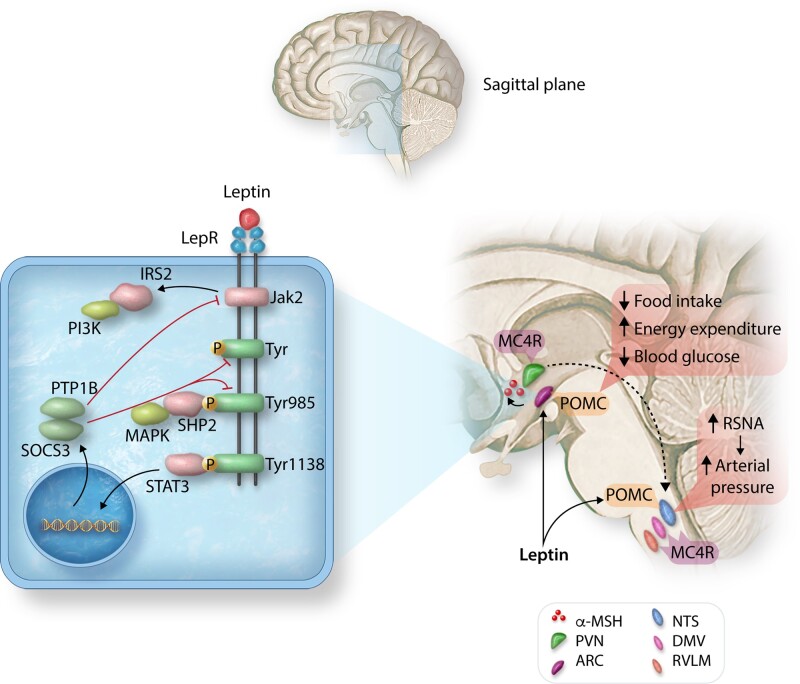
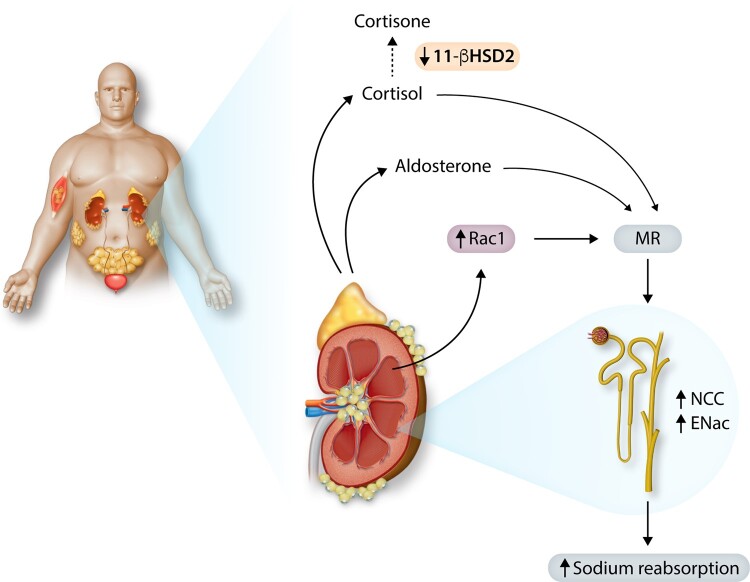
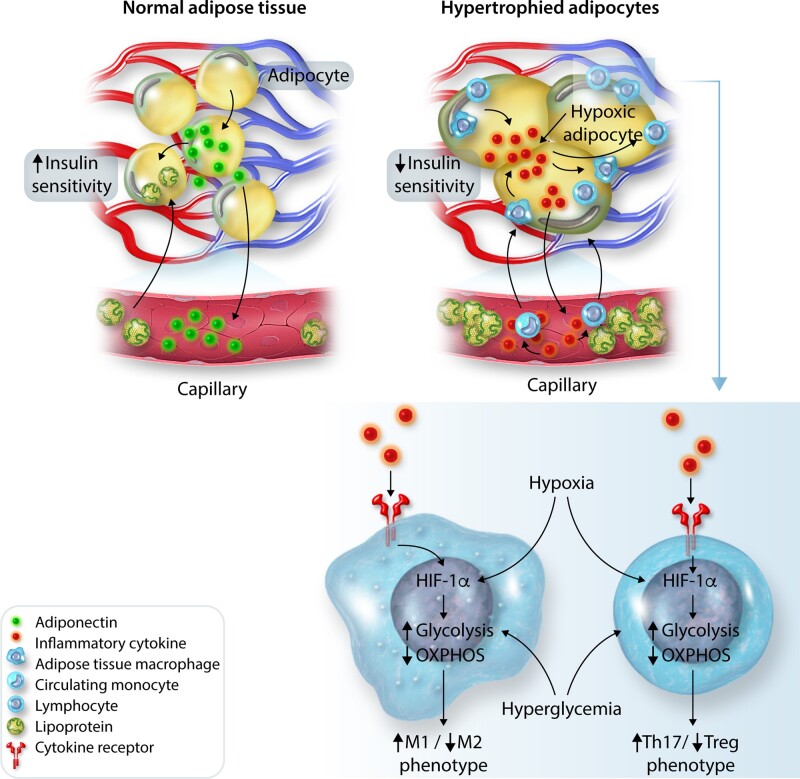
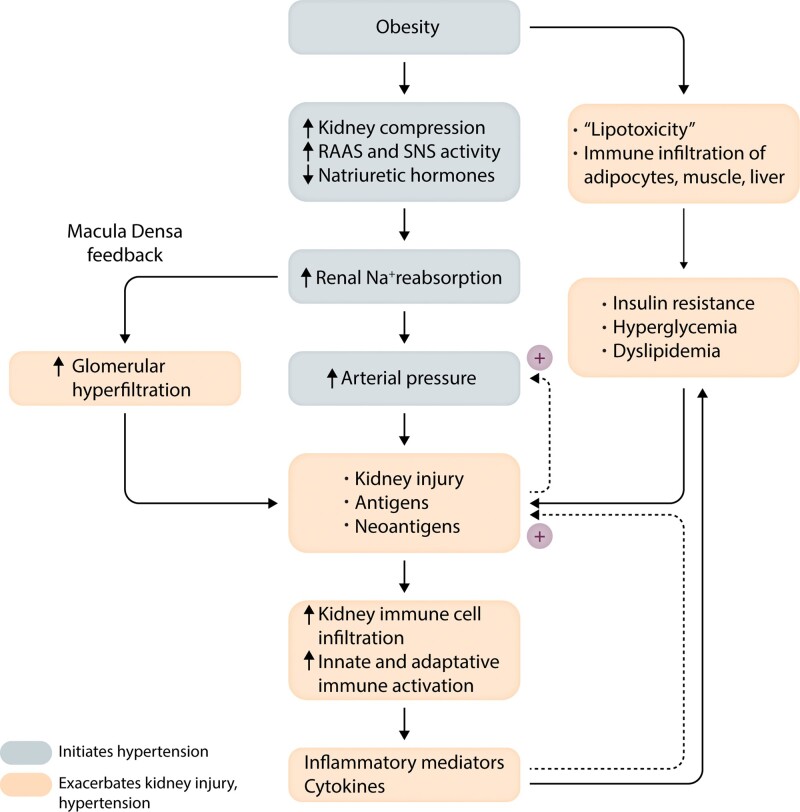
Obesity contributes 65-75% of the risk for human primary (essential) hypertension (HT) which is a major driver of cardiovascular and kidney diseases. Kidney dysfunction, associated with increased renal sodium reabsorption and compensatory glomerular hyperfiltration, plays a key role in initiating obesity-HT and target organ injury. Mediators of kidney dysfunction and increased blood pressure include (i) elevated renal sympathetic nerve activity (RSNA); (ii) increased antinatriuretic hormones such as angiotensin II and aldosterone; (iii) relative deficiency of natriuretic hormones; (iv) renal compression by fat in and around the kidneys; and (v) activation of innate and adaptive immune cells that invade tissues throughout the body, producing inflammatory cytokines/chemokines that contribute to vascular and target organ injury, and exacerbate HT. These neurohormonal, renal, and inflammatory mechanisms of obesity-HT are interdependent. For example, excess adiposity increases the adipocyte-derived cytokine leptin which increases RSNA by stimulating the central nervous system proopiomelanocortin-melanocortin 4 receptor pathway. Excess visceral, perirenal and renal sinus fat compress the kidneys which, along with increased RSNA, contribute to renin-angiotensin-aldosterone system activation, although obesity may also activate mineralocorticoid receptors independent of aldosterone. Prolonged obesity, HT, metabolic abnormalities, and inflammation cause progressive renal injury, making HT more resistant to therapy and often requiring multiple antihypertensive drugs and concurrent treatment of dyslipidaemia, insulin resistance, diabetes, and inflammation. More effective anti-obesity drugs are needed to prevent the cascade of cardiorenal, metabolic, and immune disorders that threaten to overwhelm health care systems as obesity prevalence continues to increase.
Keywords: Adipose; Blood pressure; Chronic kidney disease; Immune cells; Leptin; Melanocortins; Renin–angiotensin–aldosterone system; Sympathetic activity.
Published on behalf of the European Society of Cardiology. All rights reserved. © The Author(s) 2020. For permissions, please email: journals.permissions@oup.com.
Figures
References
-
- Afshin A, Reitsma MB, Murray CJL.. Health effects of overweight and obesity in 195 countries. N Engl J Med 2017;377:1496–1497. - PubMed
-
- Gregg EW, Shaw JE.. Global health effects of overweight and obesity. N Engl J Med 2017;377:80–81. - PubMed
-
- World Health Organization—Obesity and Overweight—Key Facts. https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (30 September 2020, date last accessed).
-
- Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, Long MW, Gortmaker SL.. Projected U.S. state-level prevalence of adult obesity and severe obesity. N Engl J Med 2019;381:2440–2450. - PubMed
-
- Garrison RJ, Kannel WB, Stokes J III, Castelli WP.. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Prev Med 1987;16:235–251. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
