

Fabry disease pain: patient and preclinical parallels
- PMID: 33259456
- PMCID: PMC8054551
- DOI: 10.1097/j.pain.0000000000002152
Fabry disease pain: patient and preclinical parallels
Abstract
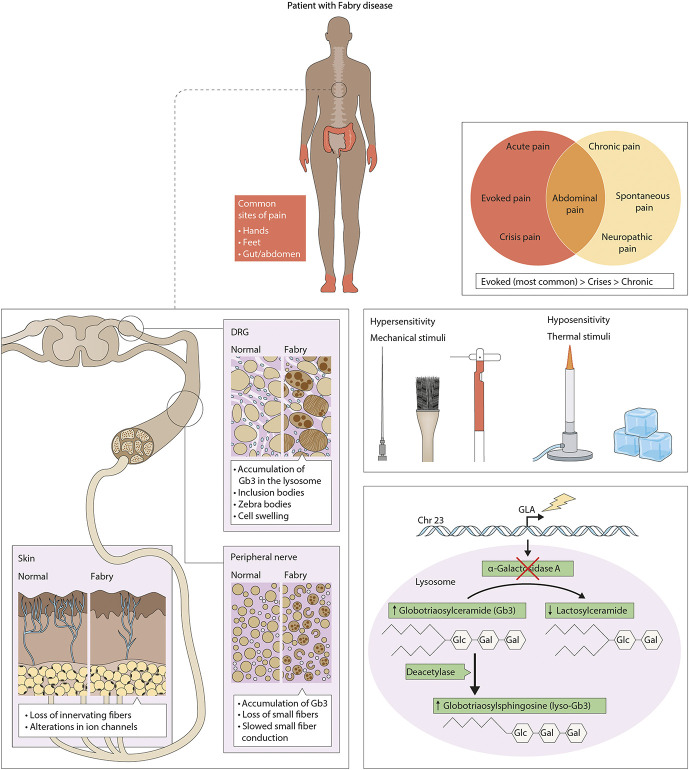
Severe neuropathic pain is a hallmark of Fabry disease, a genetic disorder caused by a deficiency in lysosomal α-galactosidase A. Pain experienced by these patients significantly impacts their quality of life and ability to perform everyday tasks. Patients with Fabry disease suffer from peripheral neuropathy, sensory abnormalities, acute pain crises, and lifelong ongoing pain. Although treatment of pain through medication and enzyme replacement therapy exists, pain persists in many of these patients. Some has been learned in the past decades regarding clinical manifestations of pain in Fabry disease and the pathological effects of α-galactosidase A insufficiency in neurons. Still, it is unclear how pain and sensory abnormalities arise in patients with Fabry disease and how these can be targeted with therapeutics. Our knowledge is limited in part due to the lack of adequate preclinical models to study the disease. This review will detail the types of pain, sensory abnormalities, influence of demographics on pain, and current strategies to treat pain experienced by patients with Fabry disease. In addition, we discuss the current knowledge of Fabry pain pathogenesis and which aspects of the disease preclinical models accurately recapitulate. Understanding the commonalities and divergences between humans and preclinical models can be used to further interrogate mechanisms causing the pain and sensory abnormalities as well as advance development of the next generation of therapeutics to treat pain in patients with Fabry disease.
Copyright © 2020 The Author(s). Published by Wolters Kluwer Health, Inc. on behalf of the International Association for the Study of Pain.
Conflict of interest statement
The authors have no conflicts of interest to declare.
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
Figures
References
-
- Albano B, Dinia L, Del Sette M, Gandolfo C, Sivori G, Finocchi C. Fabry disease in patients with migraine with aura. Neurol Sci 2010;31:167–9. - PubMed
-
- Altarescu G, Goldfarb L, Park K-Y, Kaneski C, Jeffries N, Litvak S, Nagle J, Schiffmann R. Identification of fifteen novel mutations and genotype-phenotype relationship in Fabry disease. Clin Genet 2001;60:46–51. - PubMed
-
- Amicus Therapeutics. GALAFOLD (migalastat) [package insert]. U.S. Food and Drug Administration website. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/208623lbl.pdf. Revised August 2018. Accessed 12 June 2020.
-
- Anderson LJ, Wyatt KM, Henley W, Nikolaou V, Waldek S, Hughes DA, Pastores GM, Logan S. Long-term effectiveness of enzyme replacement therapy in Fabry disease: results from the NCS-LSD cohort study. J Inherit Metab Dis 2014;37:969–78. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
