

Comparative transcriptome analysis of SARS-CoV, MERS-CoV, and SARS-CoV-2 to identify potential pathways for drug repurposing
- PMID: 33260034
- PMCID: PMC7683955
- DOI: 10.1016/j.compbiomed.2020.104123
Comparative transcriptome analysis of SARS-CoV, MERS-CoV, and SARS-CoV-2 to identify potential pathways for drug repurposing
Abstract
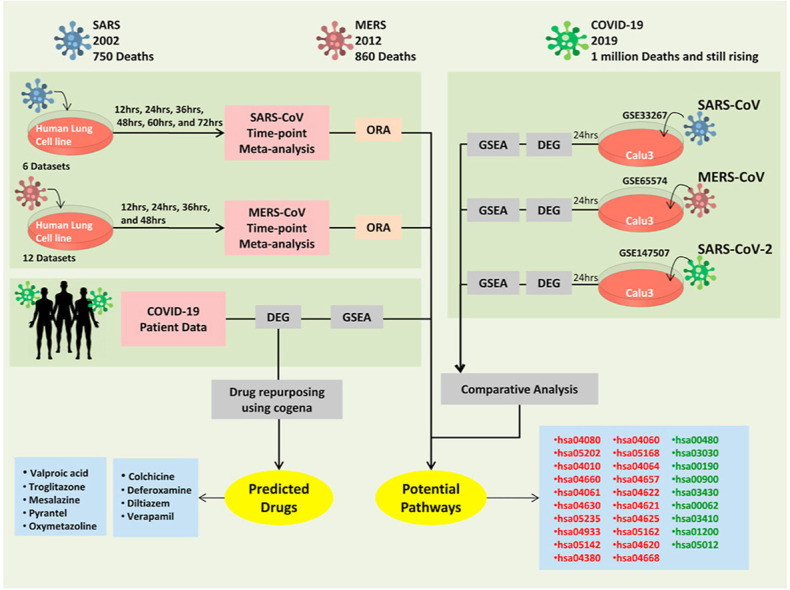
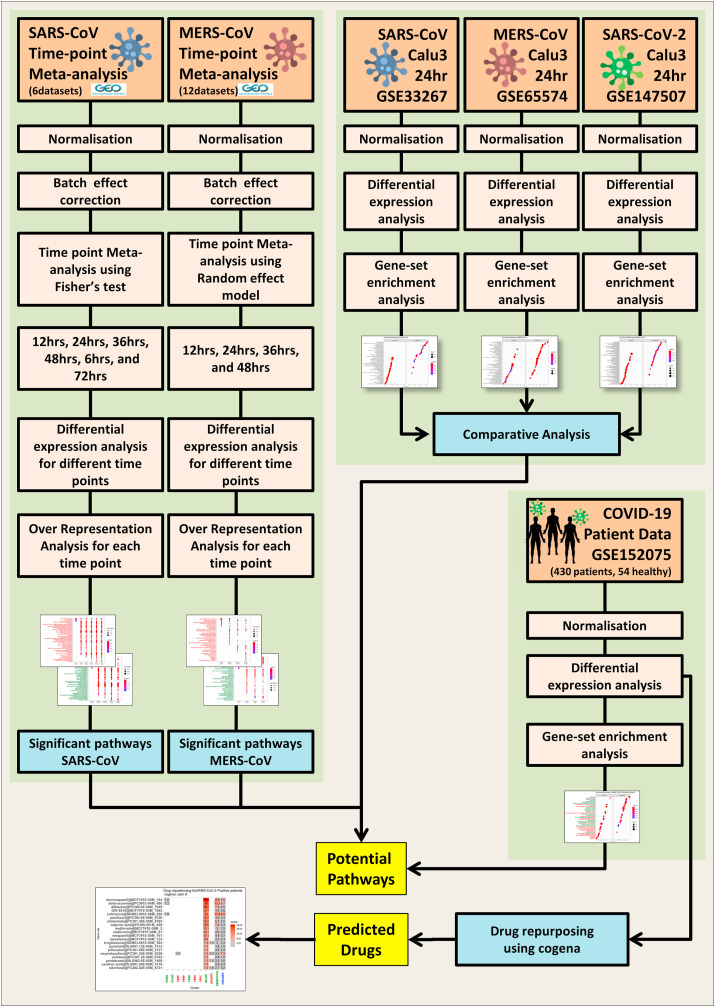
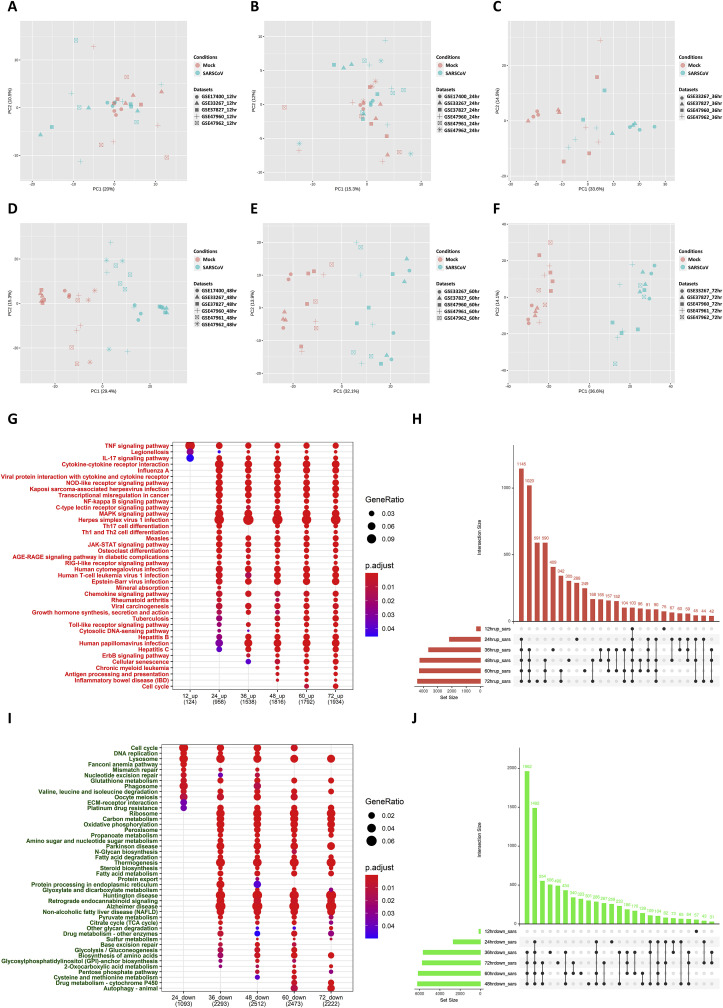
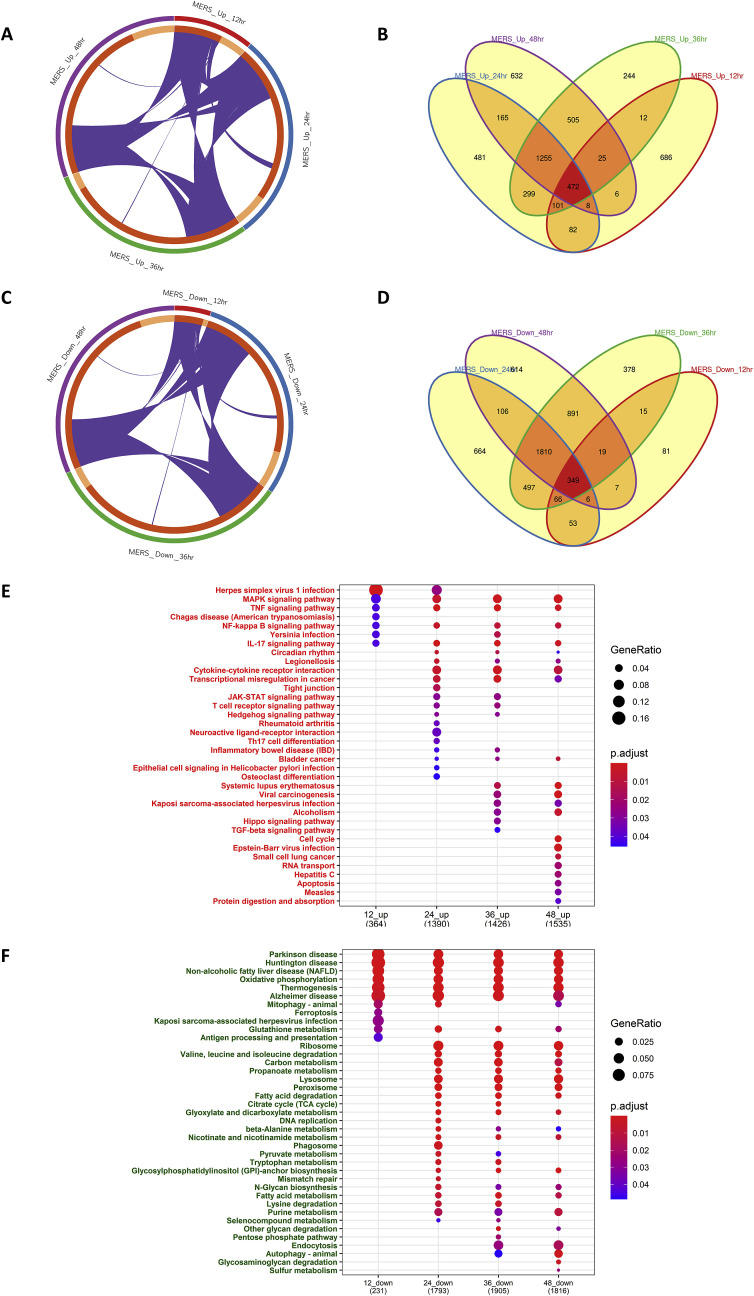
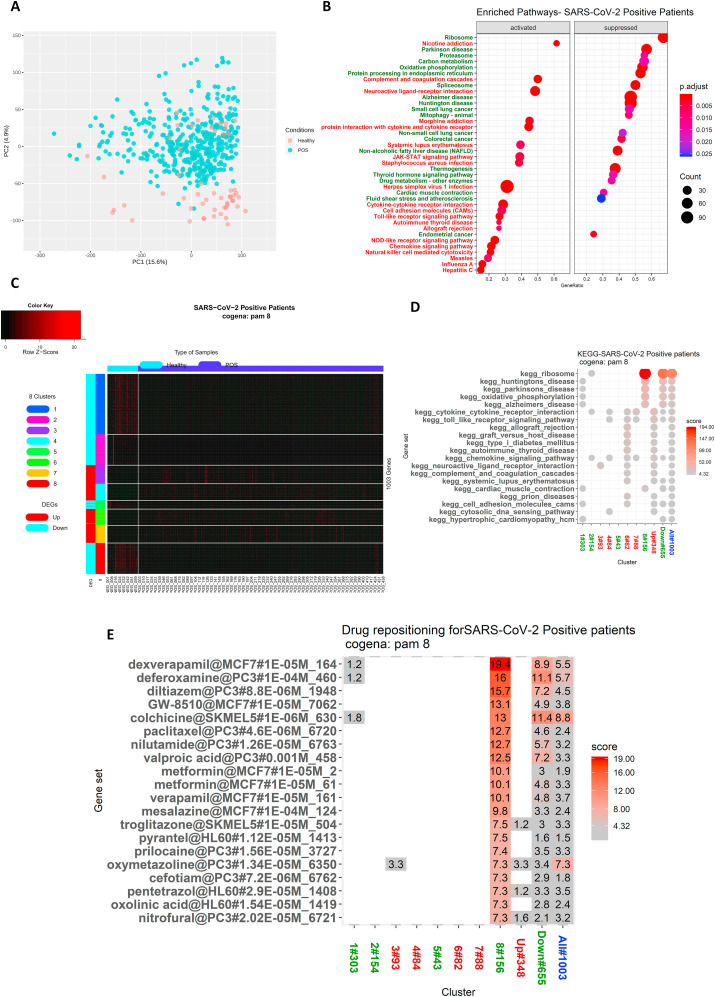
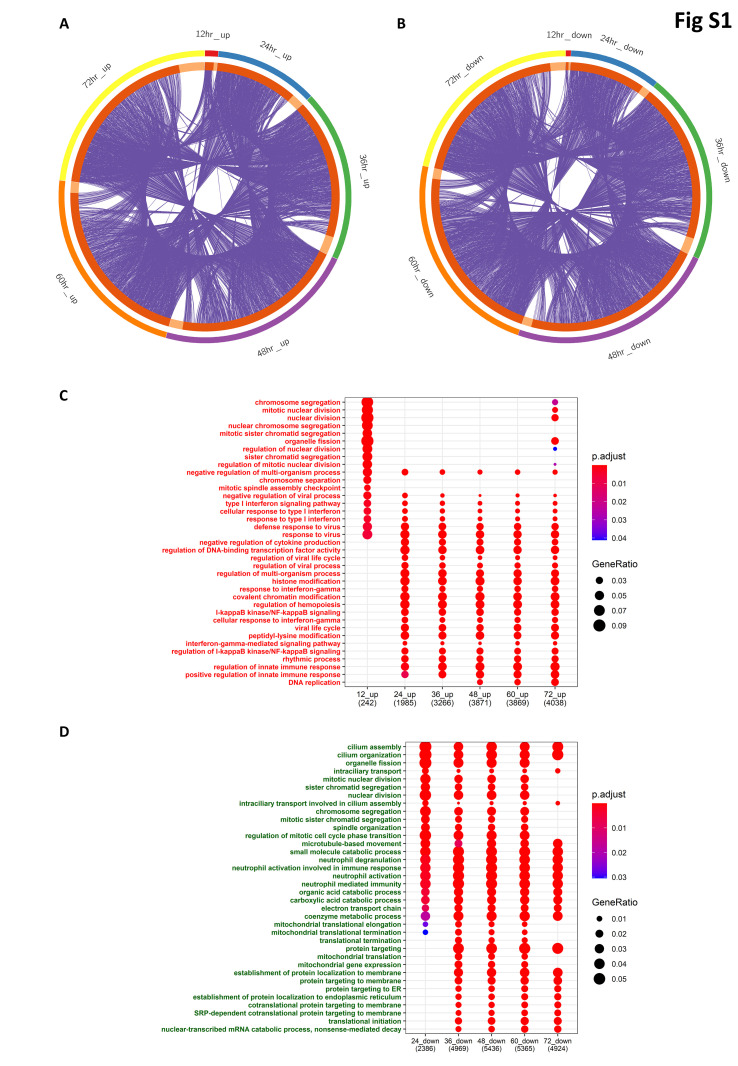
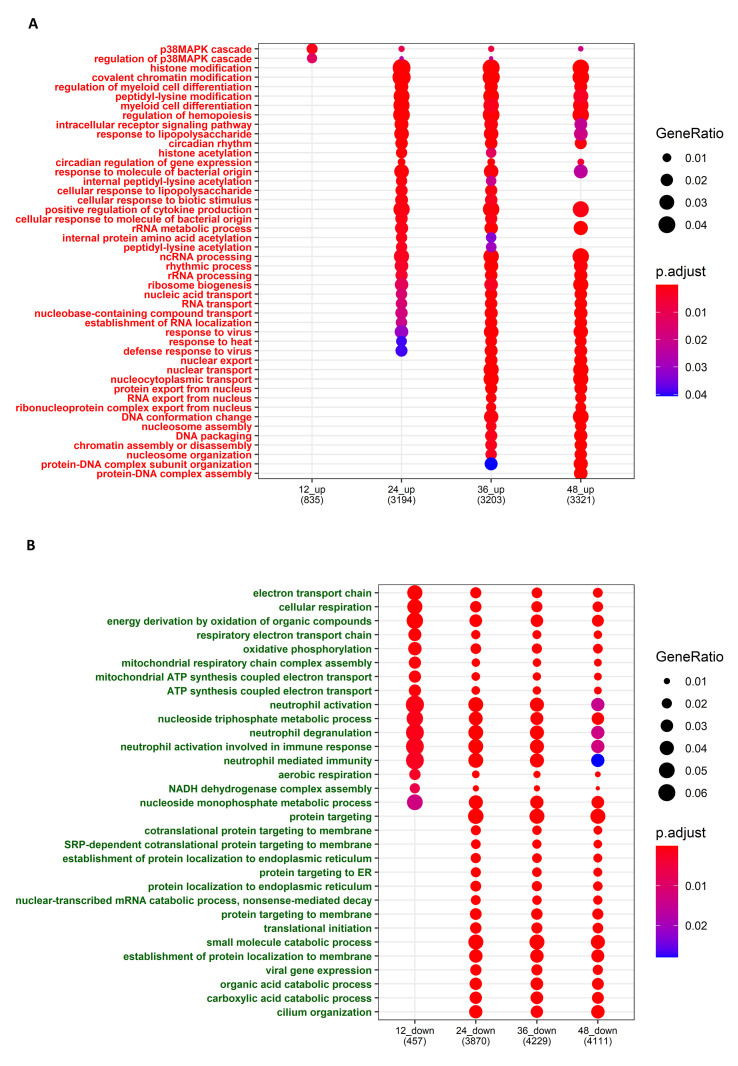
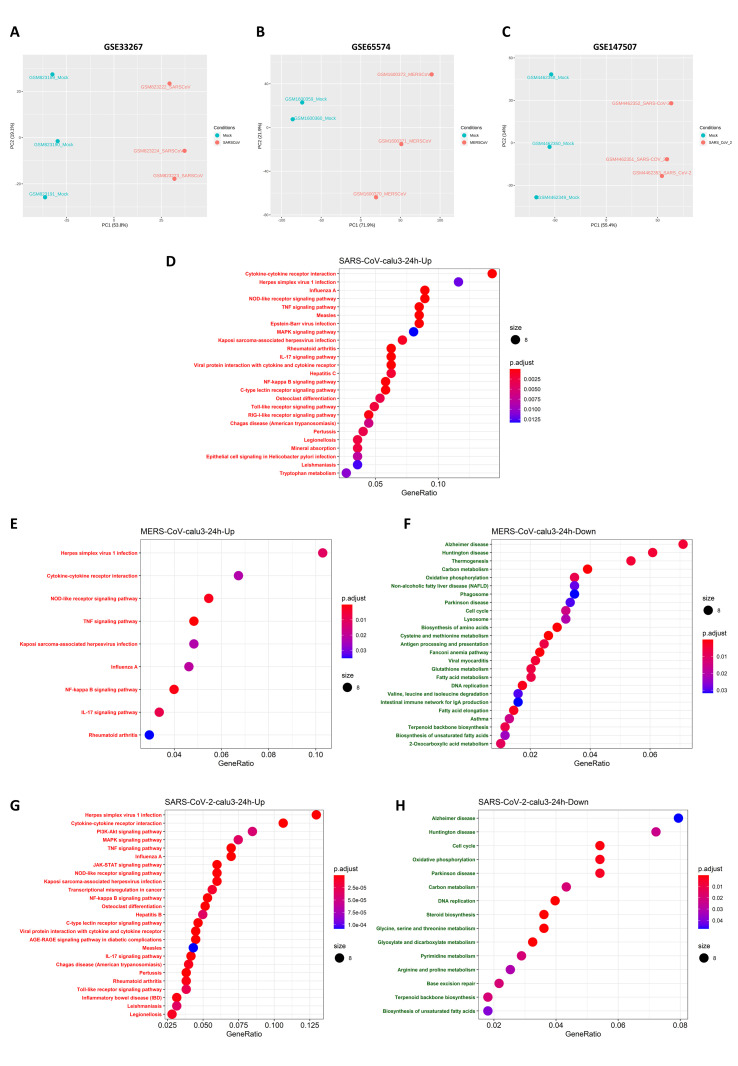
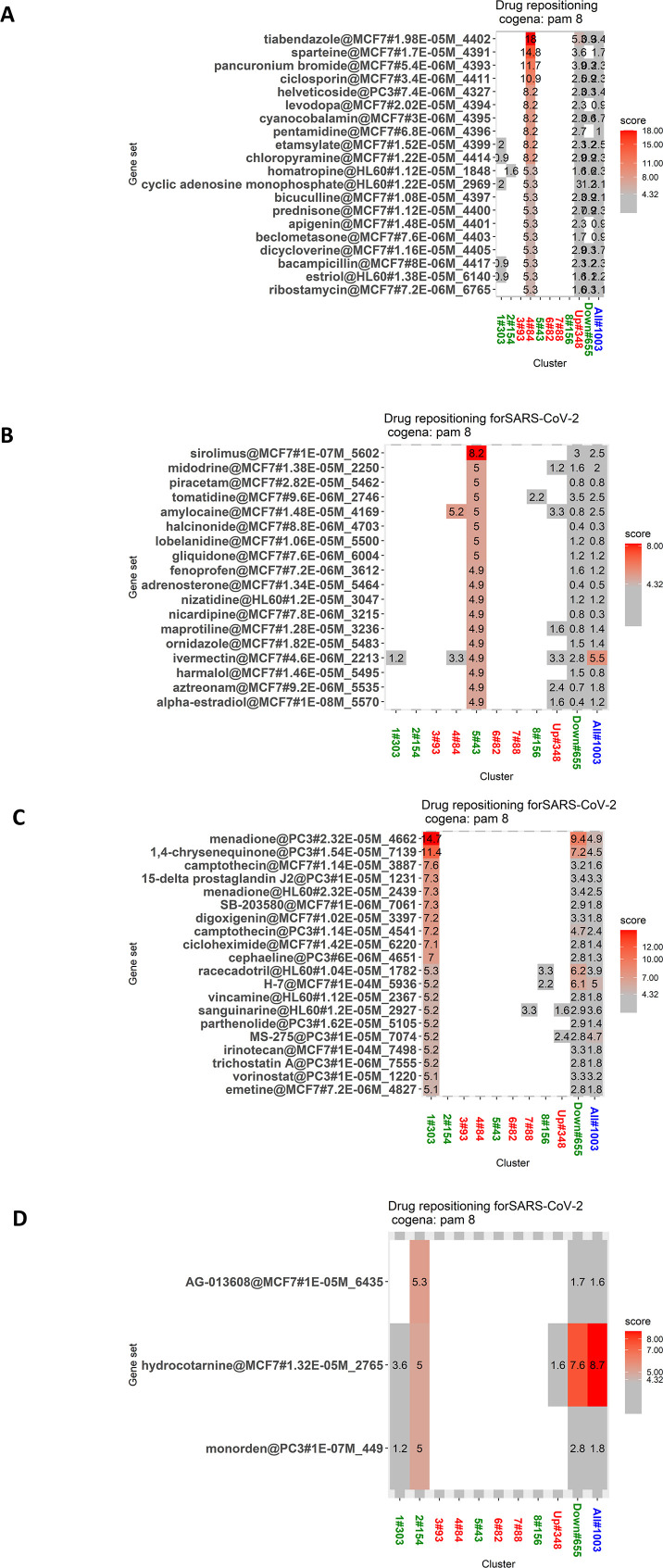
The ongoing COVID-19 pandemic caused by the coronavirus, SARS-CoV-2, has already caused in excess of 1.25 million deaths worldwide, and the number is increasing. Knowledge of the host transcriptional response against this virus and how the pathways are activated or suppressed compared to other human coronaviruses (SARS-CoV, MERS-CoV) that caused outbreaks previously can help in the identification of potential drugs for the treatment of COVID-19. Hence, we used time point meta-analysis to investigate available SARS-CoV and MERS-CoV in-vitro transcriptome datasets in order to identify the significant genes and pathways that are dysregulated at each time point. The subsequent over-representation analysis (ORA) revealed that several pathways are significantly dysregulated at each time point after both SARS-CoV and MERS-CoV infection. We also performed gene set enrichment analyses of SARS-CoV and MERS-CoV with that of SARS-CoV-2 at the same time point and cell line, the results of which revealed that common pathways are activated and suppressed in all three coronaviruses. Furthermore, an analysis of an in-vivo transcriptomic dataset of COVID-19 patients showed that similar pathways are enriched to those identified in the earlier analyses. Based on these findings, a drug repurposing analysis was performed to identify potential drug candidates for combating COVID-19.
Keywords: Coronavirus; Drug candidates; Drug repurposing; Meta-analysis; SARS-CoV-2; Transcriptome.
Copyright © 2020 Elsevier Ltd. All rights reserved.
Conflict of interest statement
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Figures

Similar articles
-
Nucleoside Analogs and Nucleoside Precursors as Drugs in the Fight against SARS-CoV-2 and Other Coronaviruses.Molecules. 2021 Feb 13;26(4):986. doi: 10.3390/molecules26040986. Molecules. 2021. PMID: 33668428 Free PMC article. Review.
-
The lethal internal face of the coronaviruses: Kidney tropism of the SARS, MERS, and COVID19 viruses.IUBMB Life. 2021 Aug;73(8):1005-1015. doi: 10.1002/iub.2516. Epub 2021 Jun 30. IUBMB Life. 2021. PMID: 34118117 Free PMC article. Review.
-
Elucidating the Drug Repurposing Spectra of COVID-19 with its Analogues SARS and MERS.Mini Rev Med Chem. 2021;21(20):3191-3202. doi: 10.2174/1389557521666210225114733. Mini Rev Med Chem. 2021. PMID: 33632095 Review.
-
Comprehensive Structural and Molecular Comparison of Spike Proteins of SARS-CoV-2, SARS-CoV and MERS-CoV, and Their Interactions with ACE2.Cells. 2020 Dec 8;9(12):2638. doi: 10.3390/cells9122638. Cells. 2020. PMID: 33302501 Free PMC article. Review.
-
H2V: a database of human genes and proteins that respond to SARS-CoV-2, SARS-CoV, and MERS-CoV infection.BMC Bioinformatics. 2021 Jan 7;22(1):18. doi: 10.1186/s12859-020-03935-2. BMC Bioinformatics. 2021. PMID: 33413085 Free PMC article.
Cited by
-
HYGIEIA: HYpothesizing the Genesis of Infectious Diseases and Epidemics through an Integrated Systems Biology Approach.Viruses. 2022 Jun 23;14(7):1373. doi: 10.3390/v14071373. Viruses. 2022. PMID: 35891354 Free PMC article.
-
Establishment of a human organoid-based evaluation system for assessing interspecies infection risk of animal-borne coronaviruses.Emerg Microbes Infect. 2024 Dec;13(1):2327368. doi: 10.1080/22221751.2024.2327368. Epub 2024 Mar 26. Emerg Microbes Infect. 2024. PMID: 38531008 Free PMC article.
-
Virus-specific and shared gene expression signatures in immune cells after vaccination in response to influenza and vaccinia stimulation.Front Immunol. 2023 Aug 4;14:1168784. doi: 10.3389/fimmu.2023.1168784. eCollection 2023. Front Immunol. 2023. PMID: 37600811 Free PMC article.
-
CompCorona: A web application for comparative transcriptome analyses of coronaviruses reveals SARS-CoV-2-specific host response.Turk J Biol. 2023 Dec 15;47(6):393-405. doi: 10.55730/1300-0152.2673. eCollection 2023. Turk J Biol. 2023. PMID: 38681774 Free PMC article.
-
SARS-CoV-2 potential drugs, drug targets, and biomarkers: a viral-host interaction network-based analysis.Sci Rep. 2022 Jul 13;12(1):11934. doi: 10.1038/s41598-022-15898-w. Sci Rep. 2022. PMID: 35831333 Free PMC article.
References
-
- Drosten C., Gunther S., Preiser W. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003;348(20):1967–1976. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
