

Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity
- PMID: 33262343
- PMCID: PMC7708979
- DOI: 10.1038/s41467-020-19918-z
Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity
Abstract
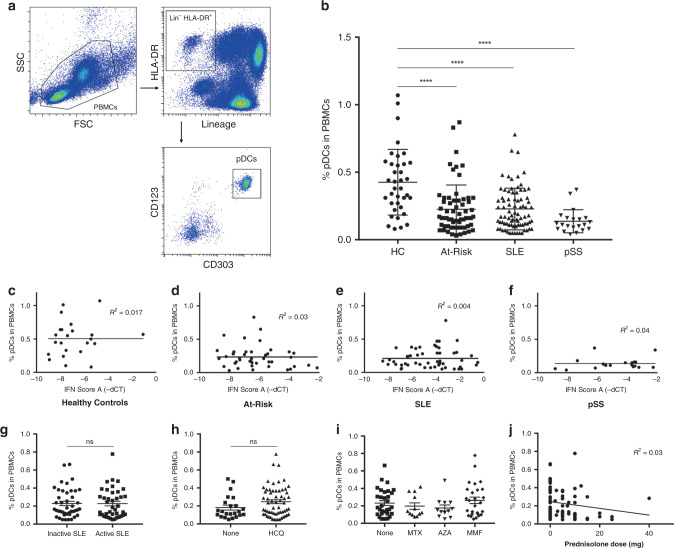
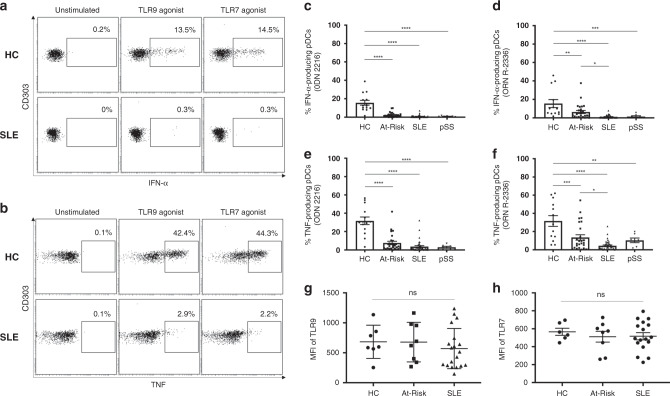
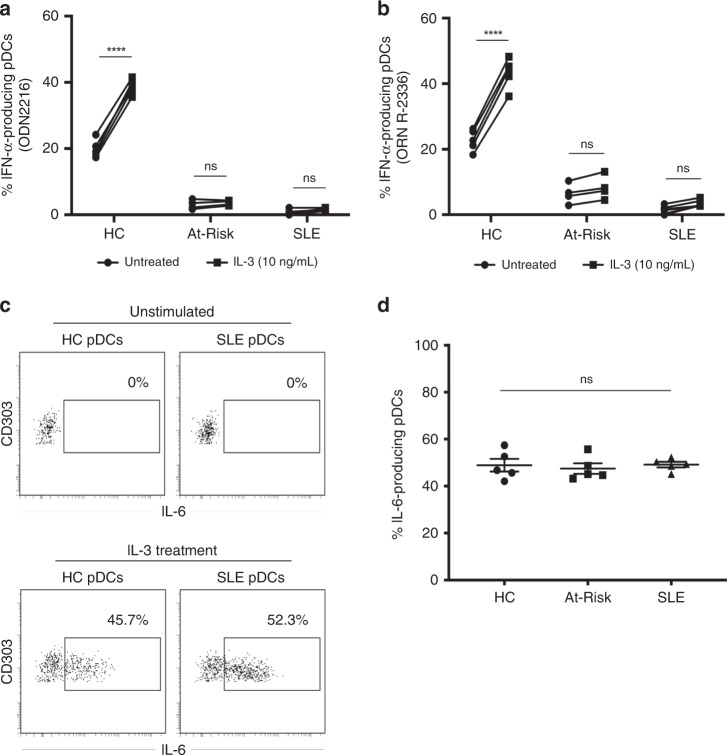
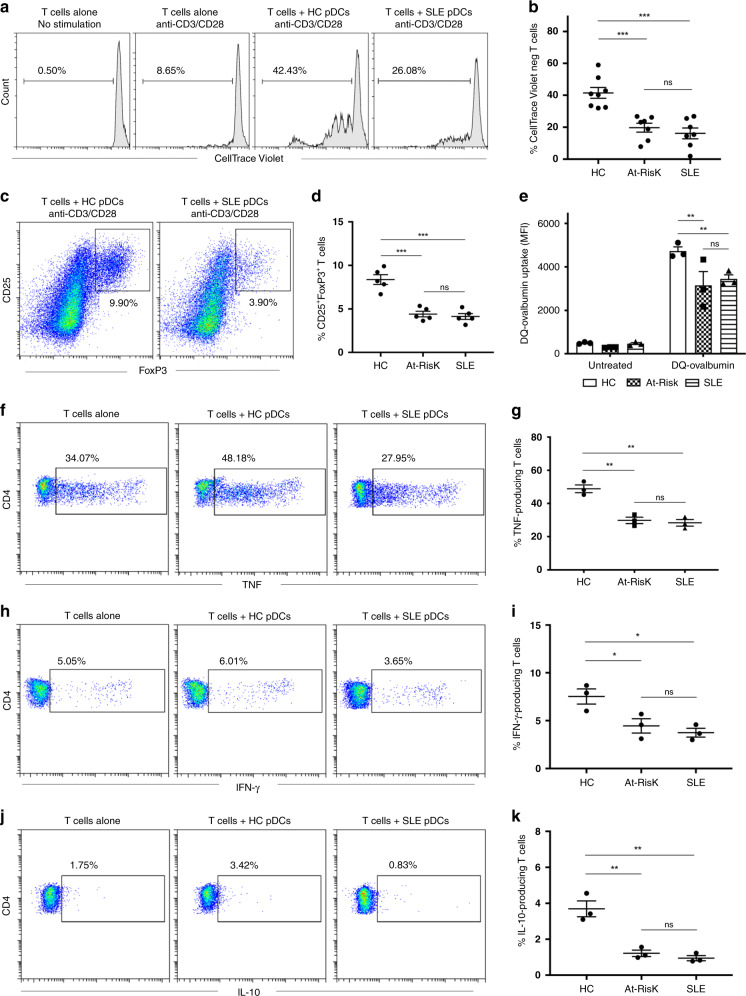
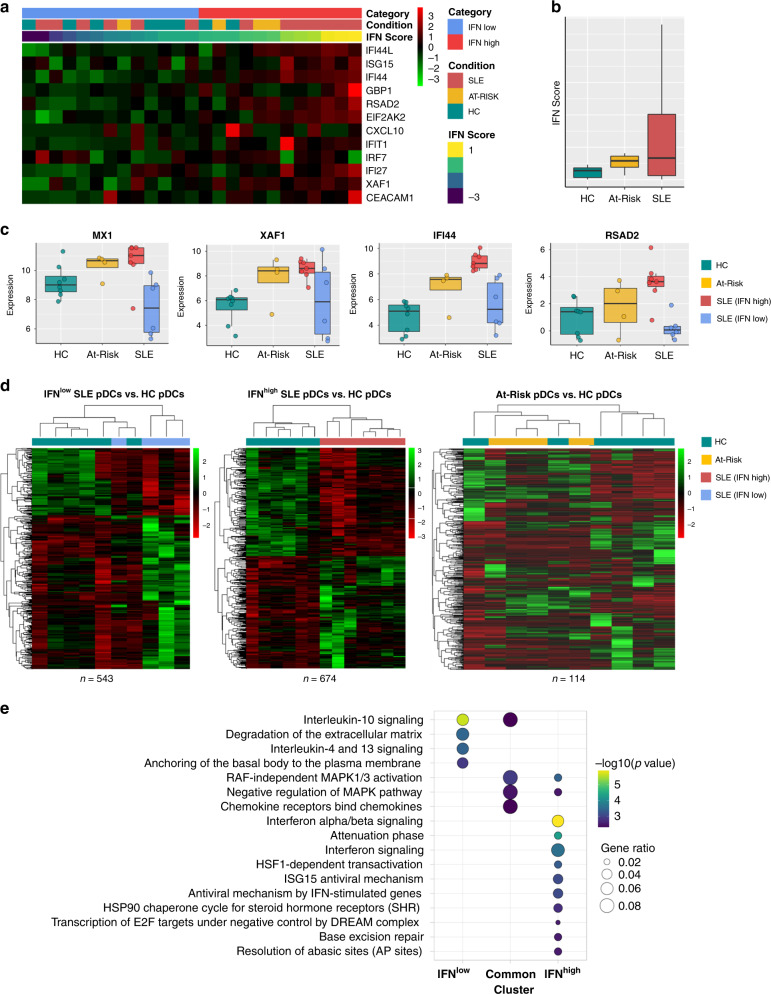
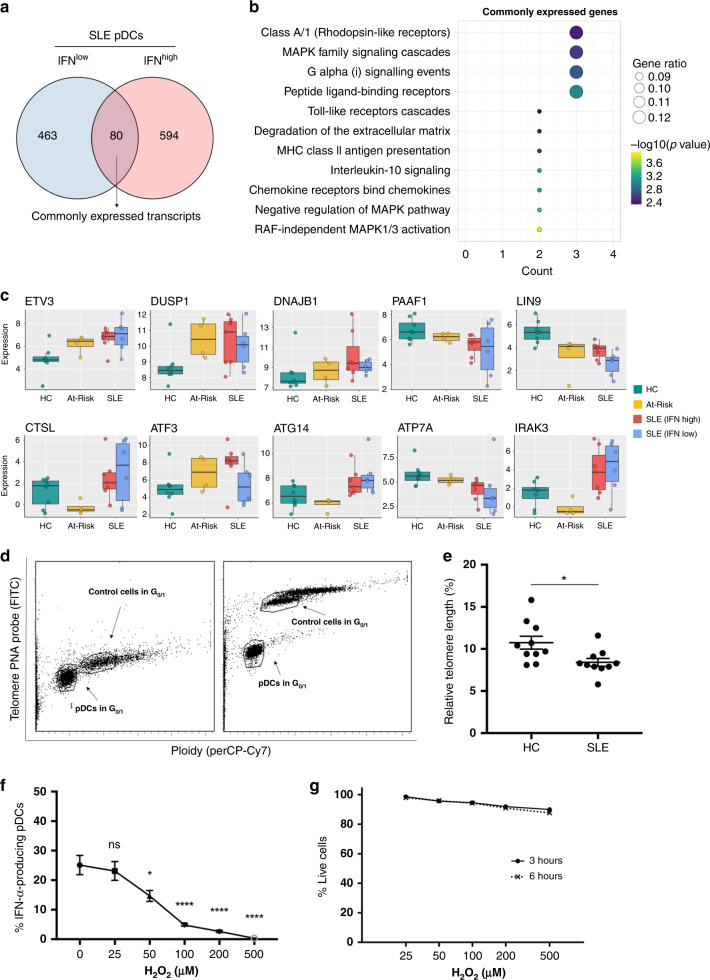
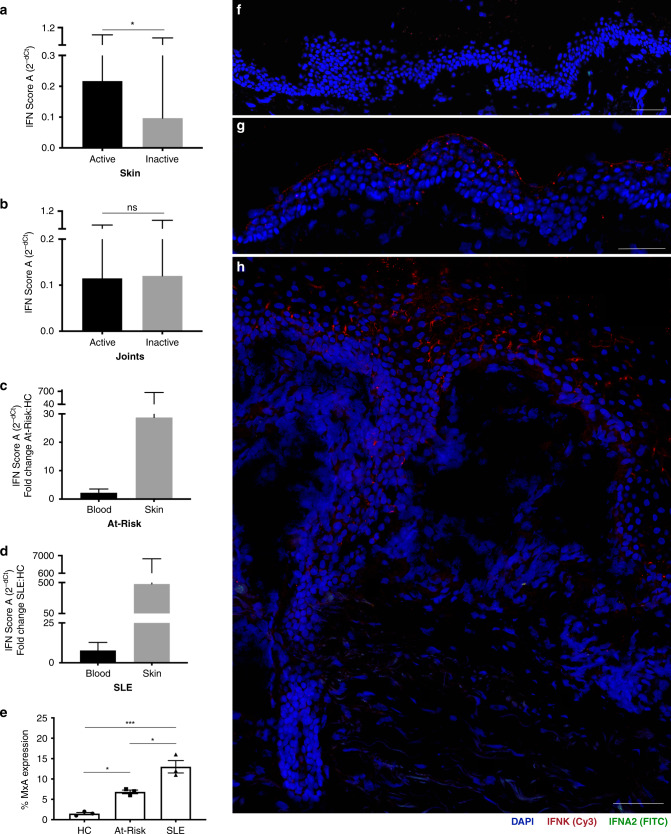
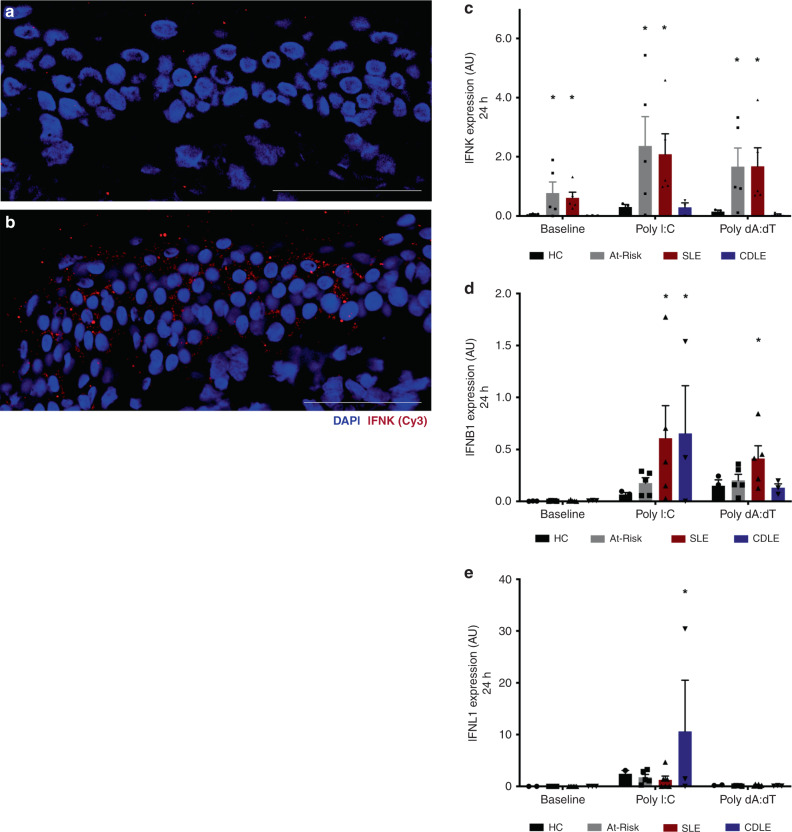
Autoimmune connective tissue diseases arise in a stepwise fashion from asymptomatic preclinical autoimmunity. Type I interferons have a crucial role in the progression to established autoimmune diseases. The cellular source and regulation in disease initiation of these cytokines is not clear, but plasmacytoid dendritic cells have been thought to contribute to excessive type I interferon production. Here, we show that in preclinical autoimmunity and established systemic lupus erythematosus, plasmacytoid dendritic cells are not effector cells, have lost capacity for Toll-like-receptor-mediated cytokine production and do not induce T cell activation, independent of disease activity and the blood interferon signature. In addition, plasmacytoid dendritic cells have a transcriptional signature indicative of cellular stress and senescence accompanied by increased telomere erosion. In preclinical autoimmunity, we show a marked enrichment of an interferon signature in the skin without infiltrating immune cells, but with interferon-κ production by keratinocytes. In conclusion, non-hematopoietic cellular sources, rather than plasmacytoid dendritic cells, are responsible for interferon production prior to clinical autoimmunity.
Conflict of interest statement
M.W. declares that they have received honoraria for educational activity and consultancy from Novartis, Janssen, Abbvie, and Celgene. P.E. declares that they have received consultancy fees from BMS, Abbott, Pfizer, MSD, Novartis, Roche, and UCB, and research grants paid to their employer from Abbott, BMS, Pfizer, MSD, and Roche. G.C.T. declares that they have received consultancy fees from Jansen and is on the Scientific Advisory Boards of Silicon Pharmaceuticals and ABPRO. E.M.V. declares that they have received consultancy fees from Roche, GSK, Lilly, and AstraZeneca, and research grants paid to his employer from Roche and AstraZeneca. The remaining authors declare no competing interests.
Figures
Comment in
-
Keratinocytes: wolves in sheep's clothing.Nat Rev Rheumatol. 2021 Feb;17(2):67. doi: 10.1038/s41584-020-00569-9. Nat Rev Rheumatol. 2021. PMID: 33402746 No abstract available.
References
-
- Psarras A, Emery P, Vital EM. Type I interferon-mediated autoimmune diseases: pathogenesis, diagnosis and targeted therapy. Rheumatology. 2017;56:1662–1675. - PubMed
-
- Niewold TB, et al. IRF5 haplotypes demonstrate diverse serological associations which predict serum interferon alpha activity and explain the majority of the genetic association with systemic lupus erythematosus. Ann. Rheum. Dis. 2012;71:463–468. doi: 10.1136/annrheumdis-2011-200463. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
