

Shift-field refinement of macromolecular atomic models
- PMID: 33263325
- PMCID: PMC7709196
- DOI: 10.1107/S2059798320013170
Shift-field refinement of macromolecular atomic models
Abstract
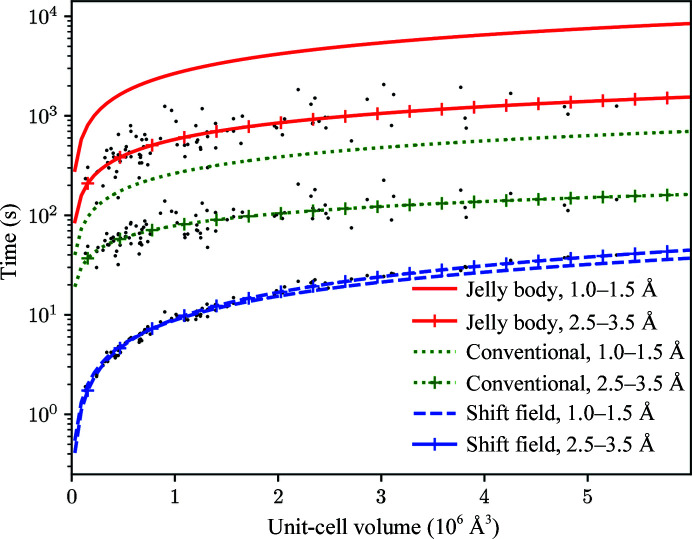
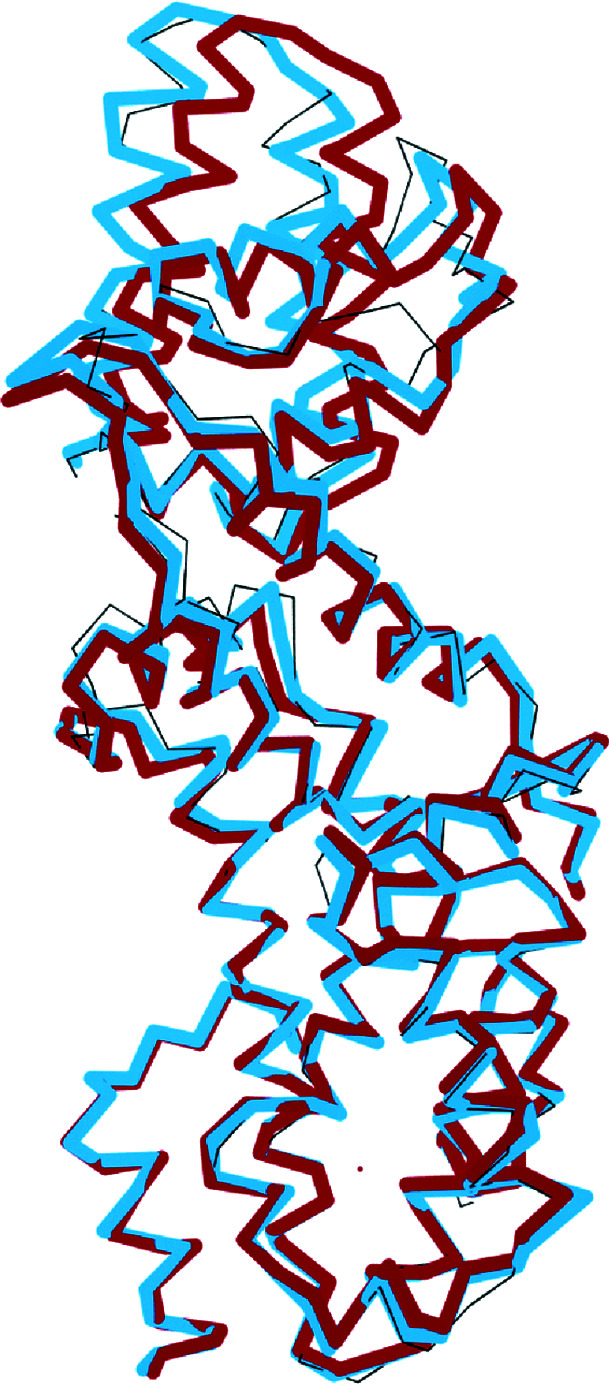
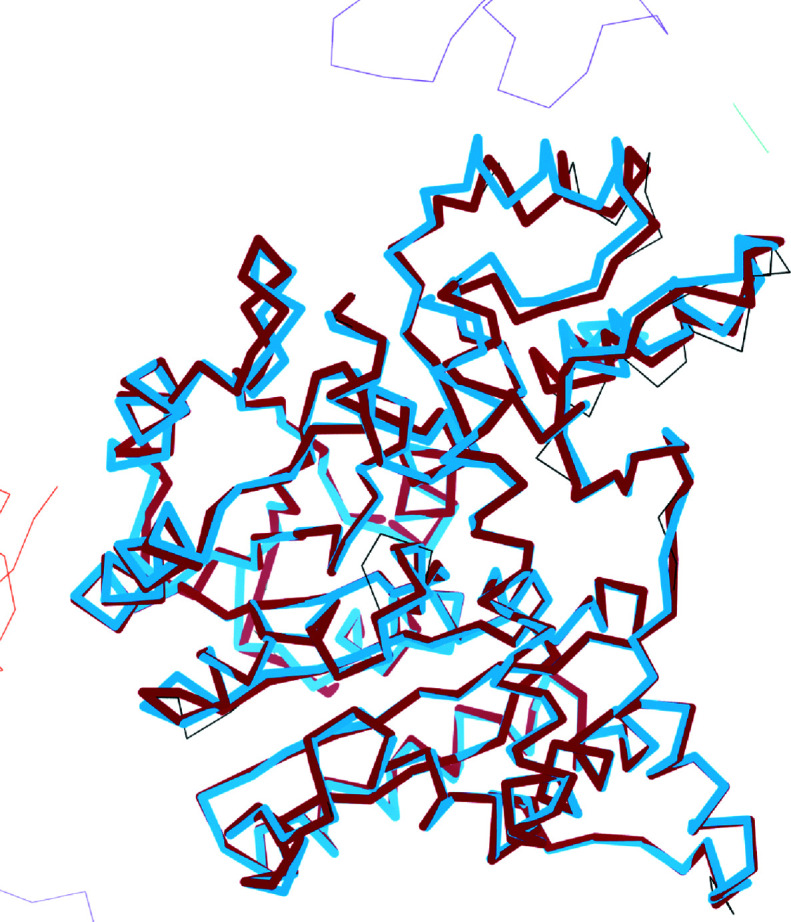
The aim of crystallographic structure solution is typically to determine an atomic model which accurately accounts for an observed diffraction pattern. A key step in this process is the refinement of the parameters of an initial model, which is most often determined by molecular replacement using another structure which is broadly similar to the structure of interest. In macromolecular crystallography, the resolution of the data is typically insufficient to determine the positional and uncertainty parameters for each individual atom, and so stereochemical information is used to supplement the observational data. Here, a new approach to refinement is evaluated in which a `shift field' is determined which describes changes to model parameters affecting whole regions of the model rather than individual atoms only, with the size of the affected region being a key parameter of the calculation which can be changed in accordance with the resolution of the data. It is demonstrated that this approach can improve the radius of convergence of the refinement calculation while also dramatically reducing the calculation time.
Keywords: computational methods; low resolution; refinement.
open access.
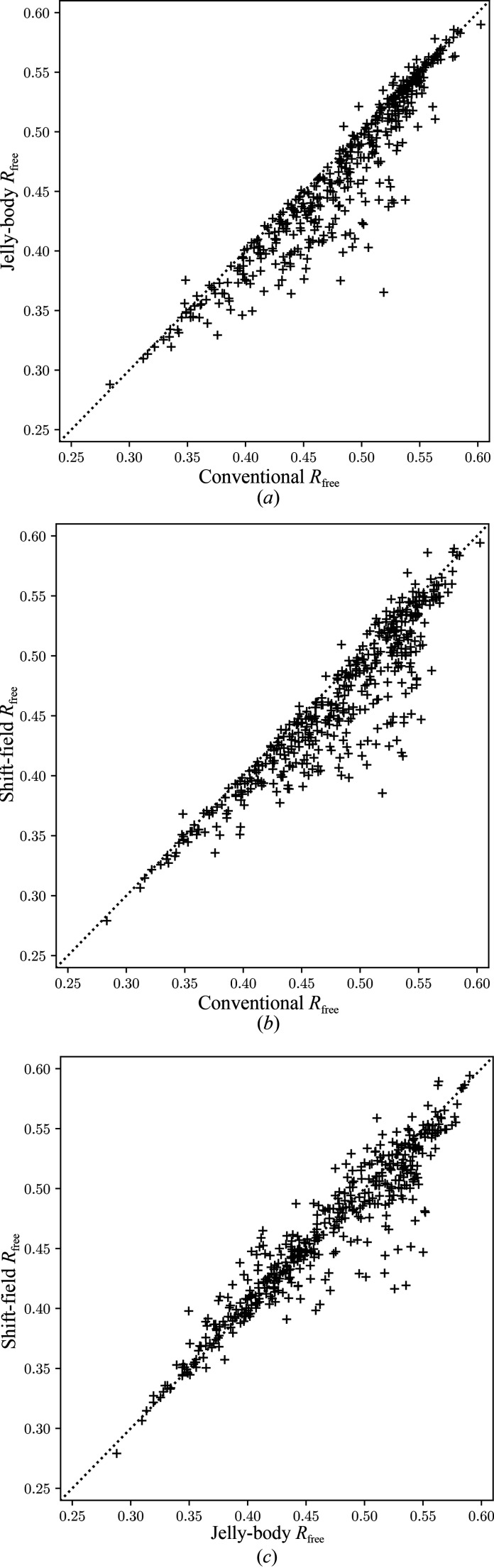
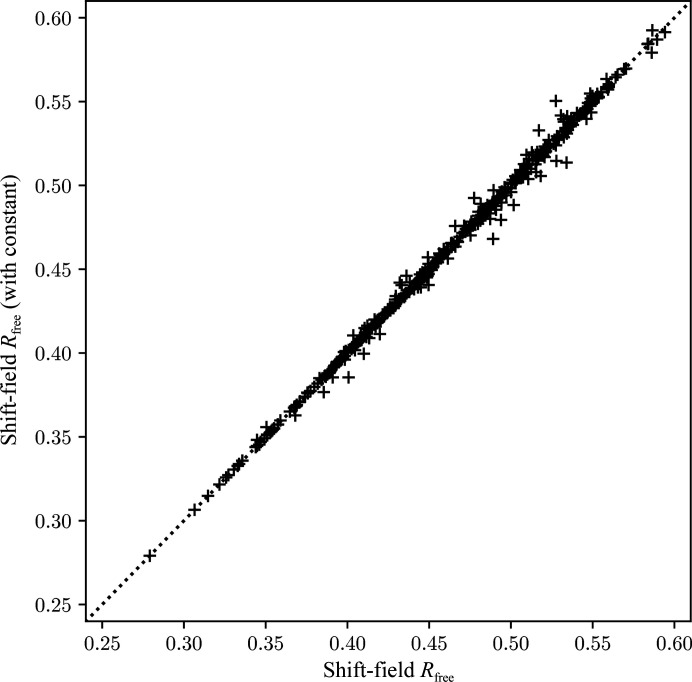
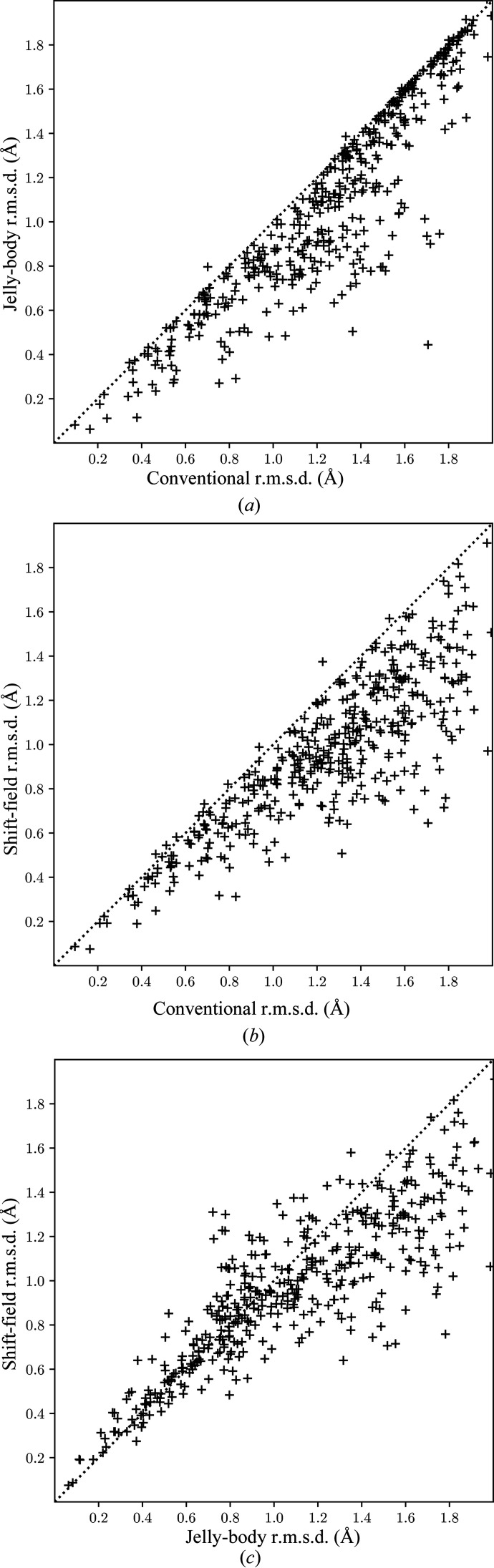
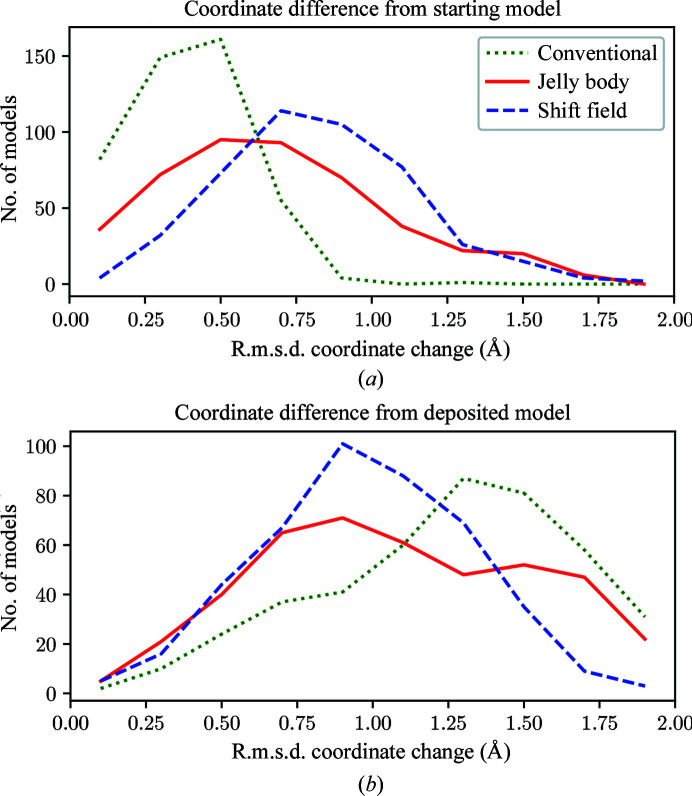
Figures
References
-
- Agarwal, R., Lifchitz, A. & Dodson, E. (1981). In Proceedings of the CCP4 Study Weekend. Refinement of Protein Structures, edited by P. A. Machin, J. W. Campbell & M. Elder. Warrington: Daresbury Laboratory.
-
- Blanc, E., Roversi, P., Vonrhein, C., Flensburg, C., Lea, S. M. & Bricogne, G. (2004). Acta Cryst. D60, 2210–2221. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
