

Developmental differences in genome replication program and origin activation
- PMID: 33264404
- PMCID: PMC7736824
- DOI: 10.1093/nar/gkaa1124
Developmental differences in genome replication program and origin activation
Abstract
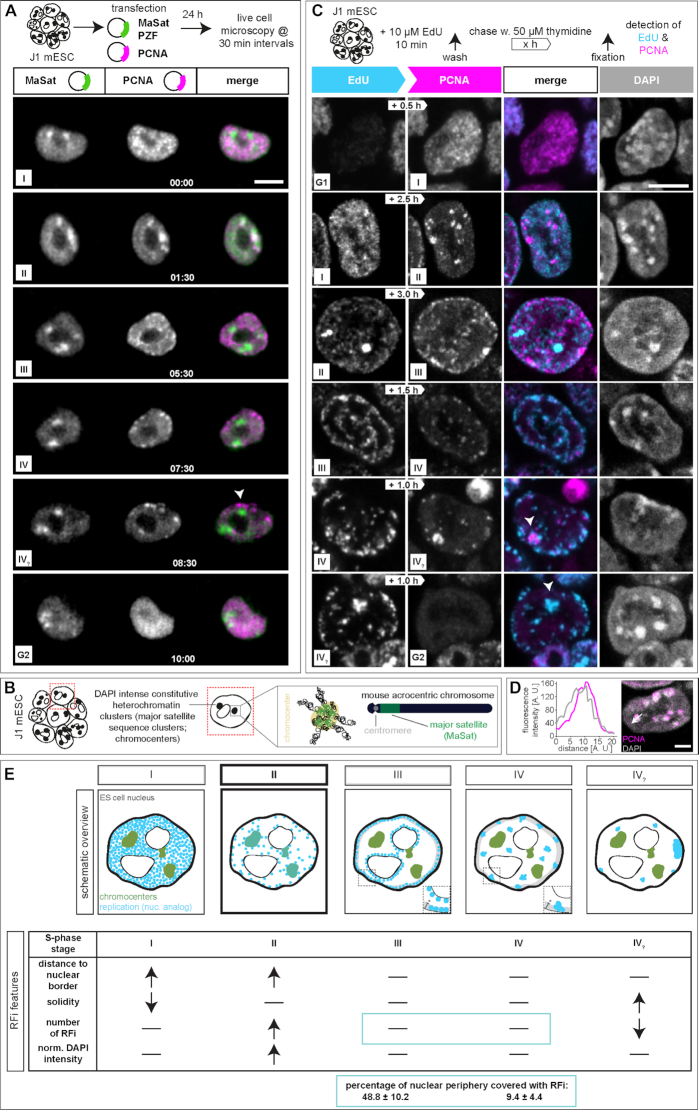
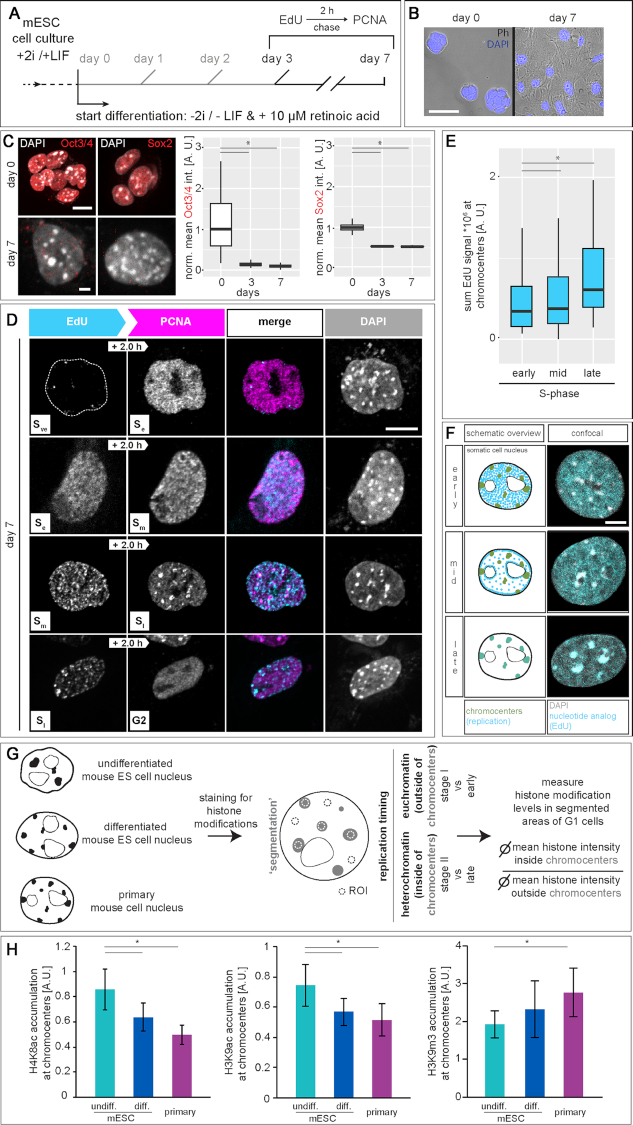
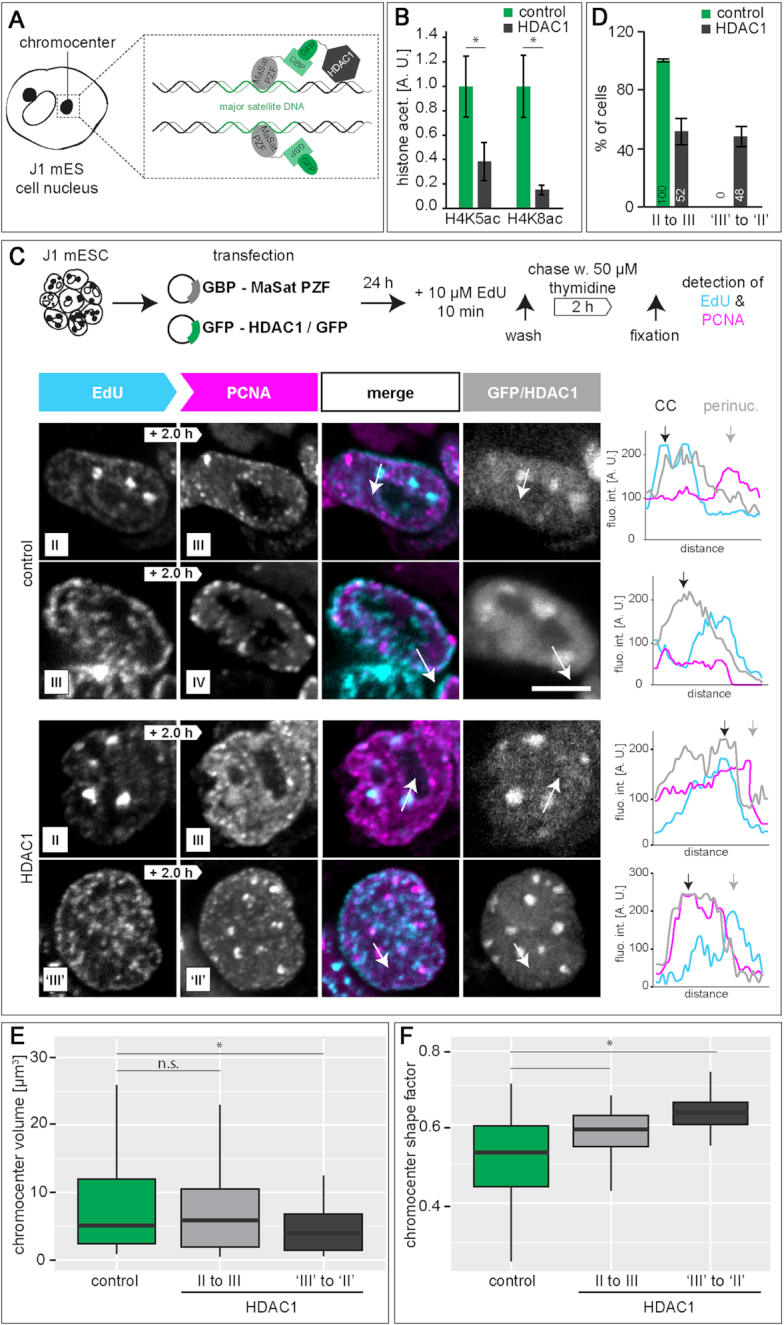
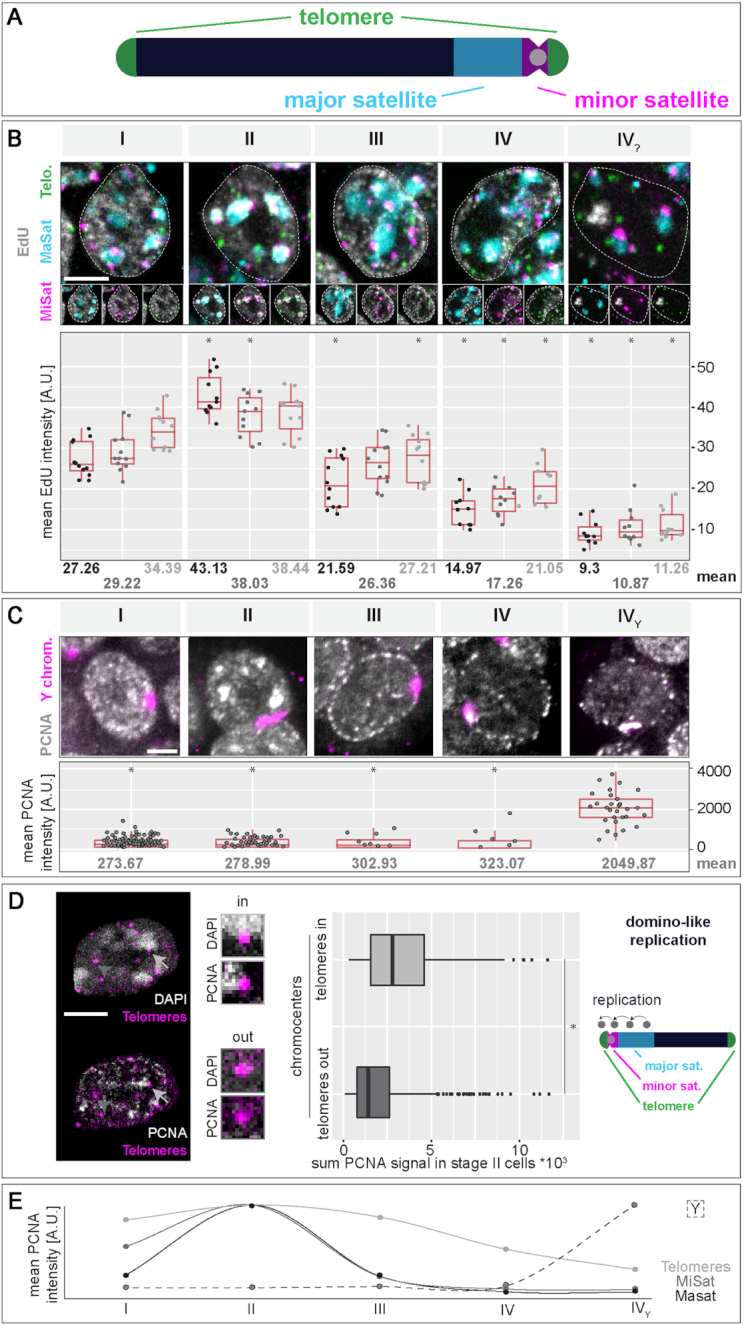
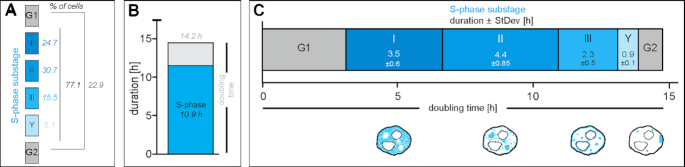
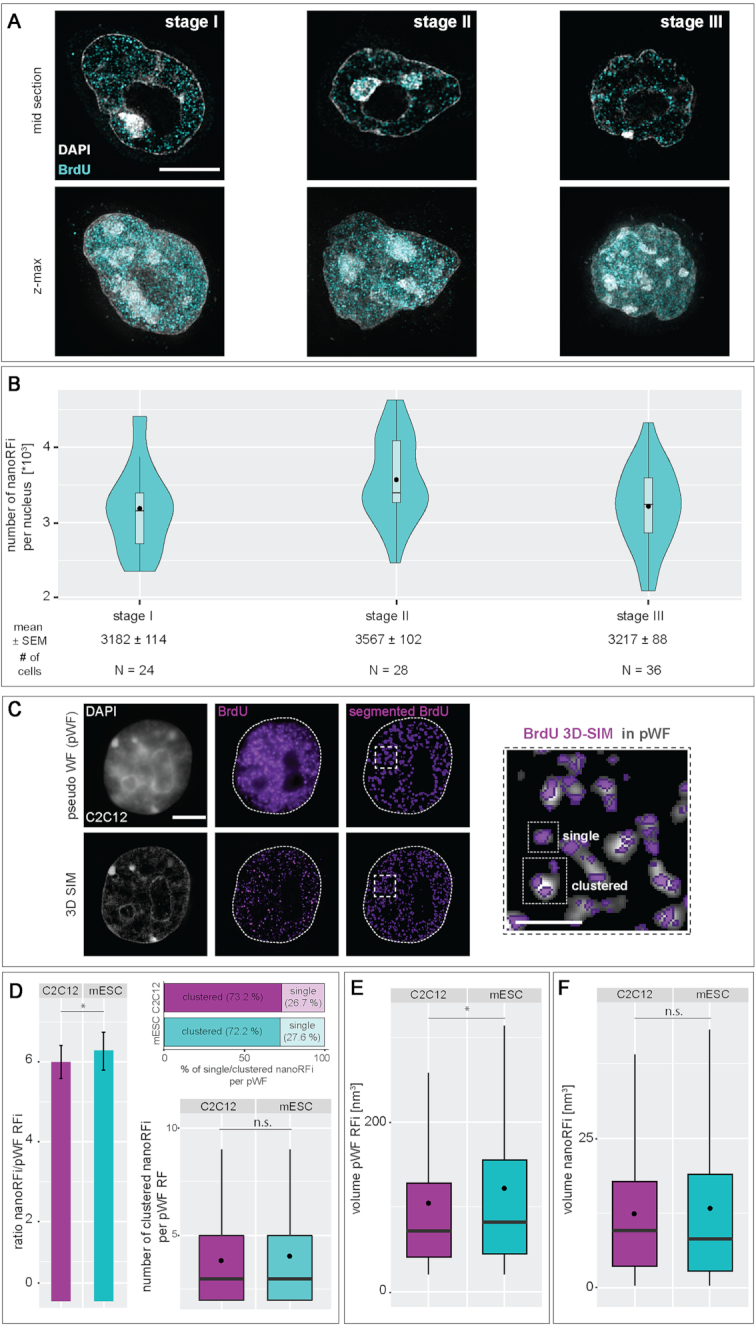
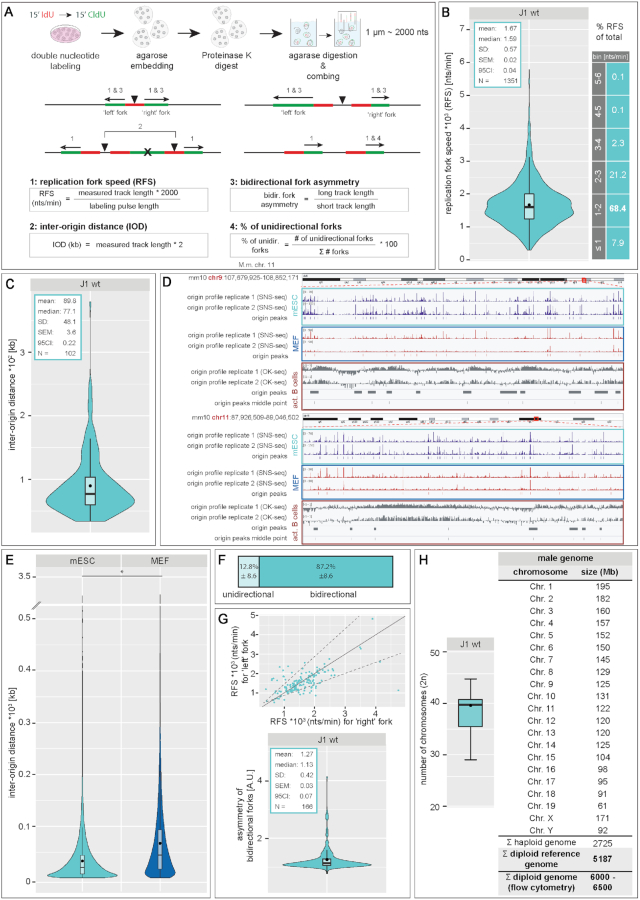
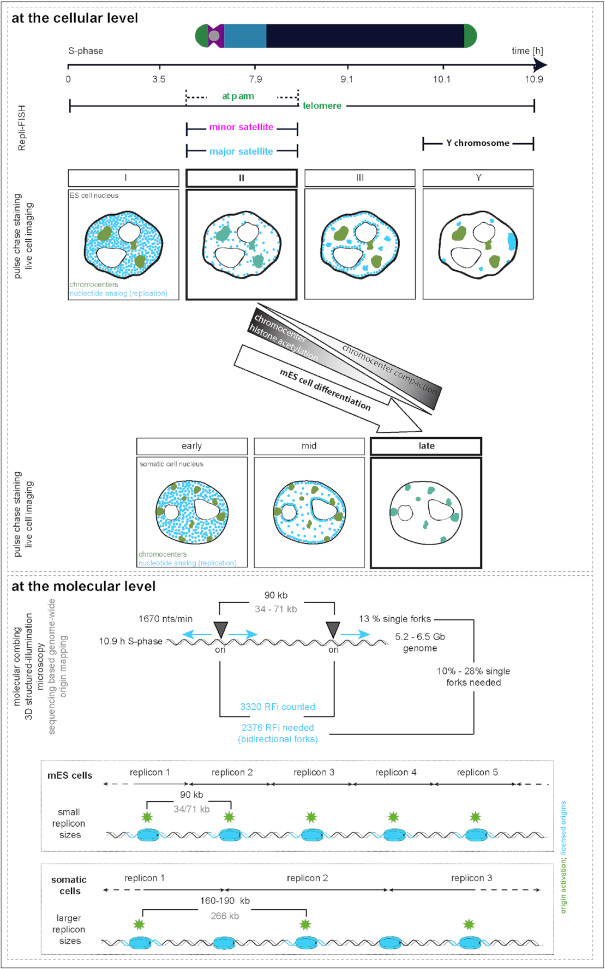
To ensure error-free duplication of all (epi)genetic information once per cell cycle, DNA replication follows a cell type and developmental stage specific spatio-temporal program. Here, we analyze the spatio-temporal DNA replication progression in (un)differentiated mouse embryonic stem (mES) cells. Whereas telomeres replicate throughout S-phase, we observe mid S-phase replication of (peri)centromeric heterochromatin in mES cells, which switches to late S-phase replication upon differentiation. This replication timing reversal correlates with and depends on an increase in condensation and a decrease in acetylation of chromatin. We further find synchronous duplication of the Y chromosome, marking the end of S-phase, irrespectively of the pluripotency state. Using a combination of single-molecule and super-resolution microscopy, we measure molecular properties of the mES cell replicon, the number of replication foci active in parallel and their spatial clustering. We conclude that each replication nanofocus in mES cells corresponds to an individual replicon, with up to one quarter representing unidirectional forks. Furthermore, with molecular combing and genome-wide origin mapping analyses, we find that mES cells activate twice as many origins spaced at half the distance than somatic cells. Altogether, our results highlight fundamental developmental differences on progression of genome replication and origin activation in pluripotent cells.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Dimitrova D.S., Gilbert D.M.. The spatial position and replication timing of chromosomal domains are both established in early G1 phase. Mol. Cell. 1999; 4:983–993. - PubMed
-
- Hyrien O., Marheineke K., Goldar A.. Paradoxes of eukaryotic DNA replication: MCM proteins and the random completion problem. BioEssays. 2003; 25:116–125. - PubMed
-
- Fragkos M., Ganier O., Coulombe P., Mechali M.. DNA replication origin activation in space and time. Nat. Rev. Mol. Cell Biol. 2015; 16:360–374. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
