

Opposing Roles of Type I Interferons in Cancer Immunity
- PMID: 33264572
- PMCID: PMC8063563
- DOI: 10.1146/annurev-pathol-031920-093932
Opposing Roles of Type I Interferons in Cancer Immunity
Abstract
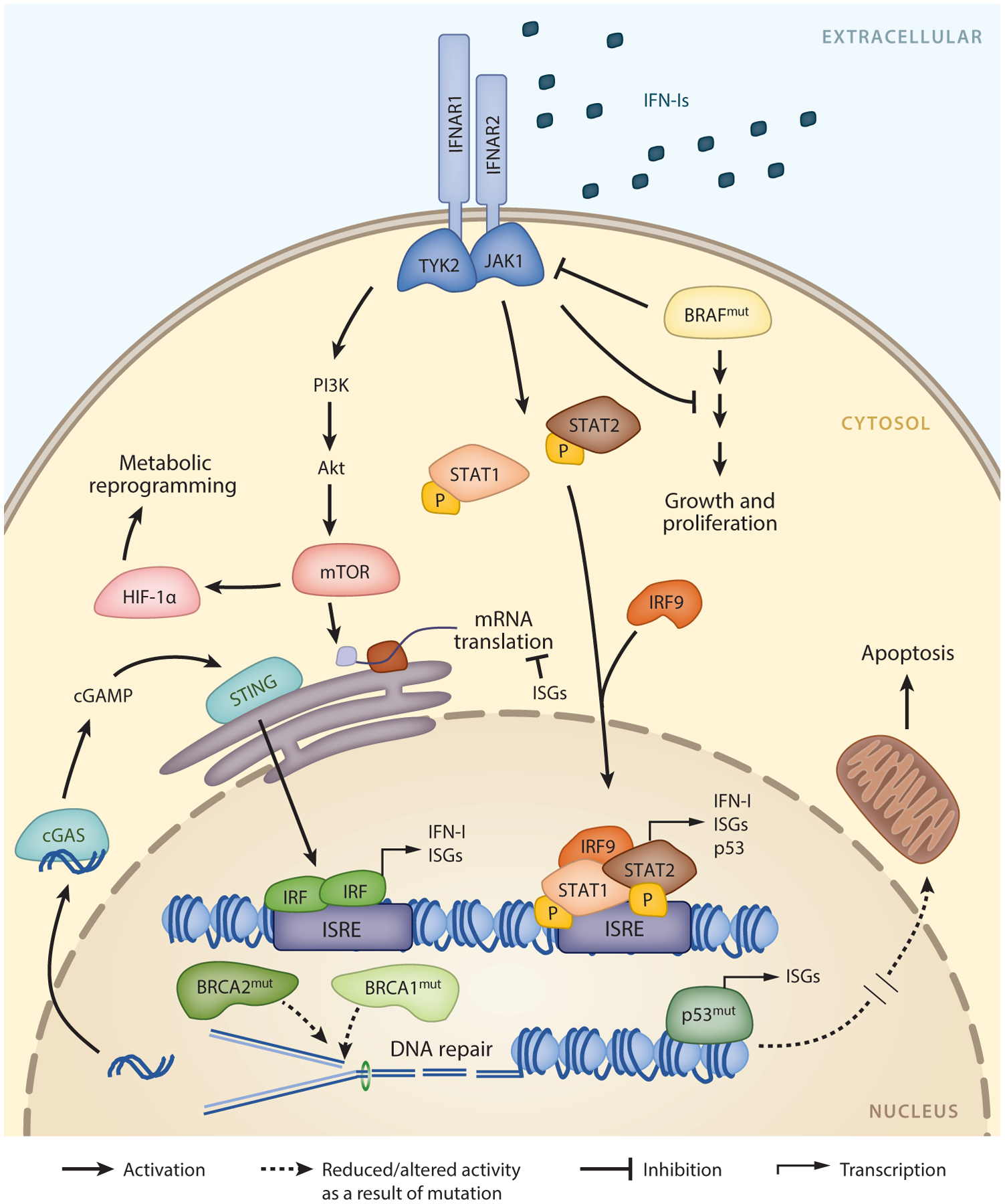
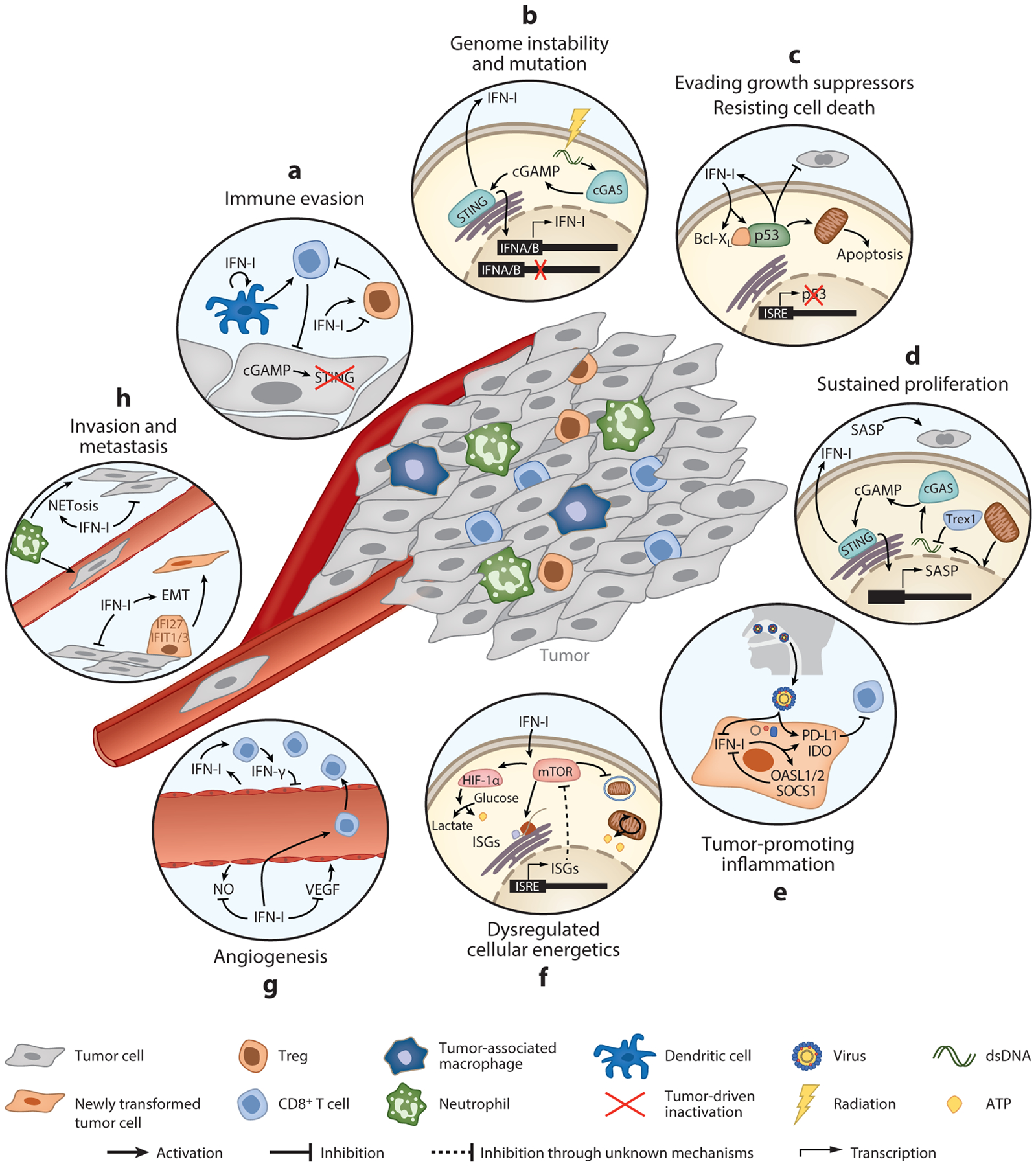
The immune system is tasked with identifying malignant cells to eliminate or prevent cancer spread. This involves a complex orchestration of many immune cell types that together recognize different aspects of tumor transformation and growth. In response, tumors have developed mechanisms to circumvent immune attack. Type I interferons (IFN-Is) are a class of proinflammatory cytokines produced in response to viruses and other environmental stressors. IFN-Is are also emerging as essential drivers of antitumor immunity, potently stimulating the ability of immune cells to eliminate tumor cells. However, a more complicated role for IFN-Is has arisen, as prolonged stimulation can promote feedback inhibitory mechanisms that contribute to immune exhaustion and other deleterious effects that directly or indirectly permit cancer cells to escape immune clearance. We review the fundamental and opposing functions of IFN-Is that modulate tumor growth and impact immune function and ultimately how these functions can be harnessed for the design of new cancer therapies.
Keywords: IFN-I; IFN-stimulated gene; ISG; cancer immunology; hallmarks of cancer; immunotherapy; type I interferon.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
