

Rare Variant Burden Analysis within Enhancers Identifies CAV1 as an ALS Risk Gene
- PMID: 33264630
- PMCID: PMC7710676
- DOI: 10.1016/j.celrep.2020.108456
Rare Variant Burden Analysis within Enhancers Identifies CAV1 as an ALS Risk Gene
Erratum in
-
Rare variant burden analysis within enhancers identifies CAV1 as an ALS risk gene.Cell Rep. 2021 Feb 2;34(5):108730. doi: 10.1016/j.celrep.2021.108730. Cell Rep. 2021. PMID: 33535055 Free PMC article. No abstract available.
Abstract
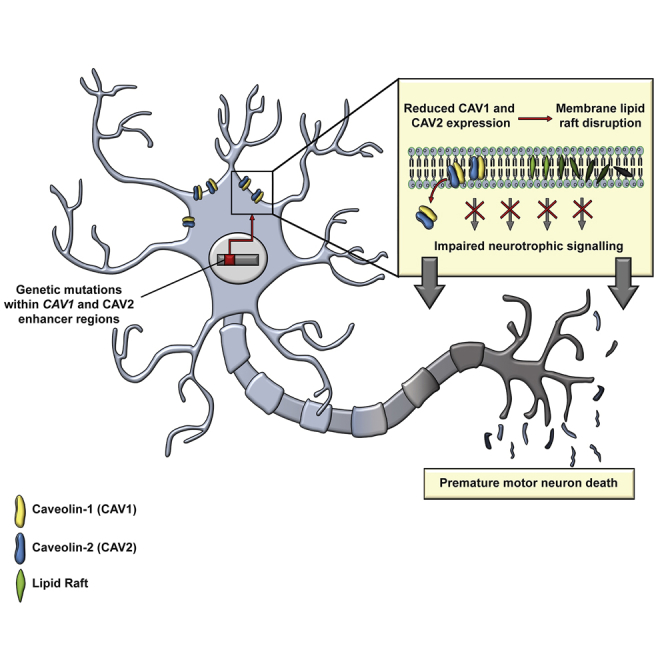
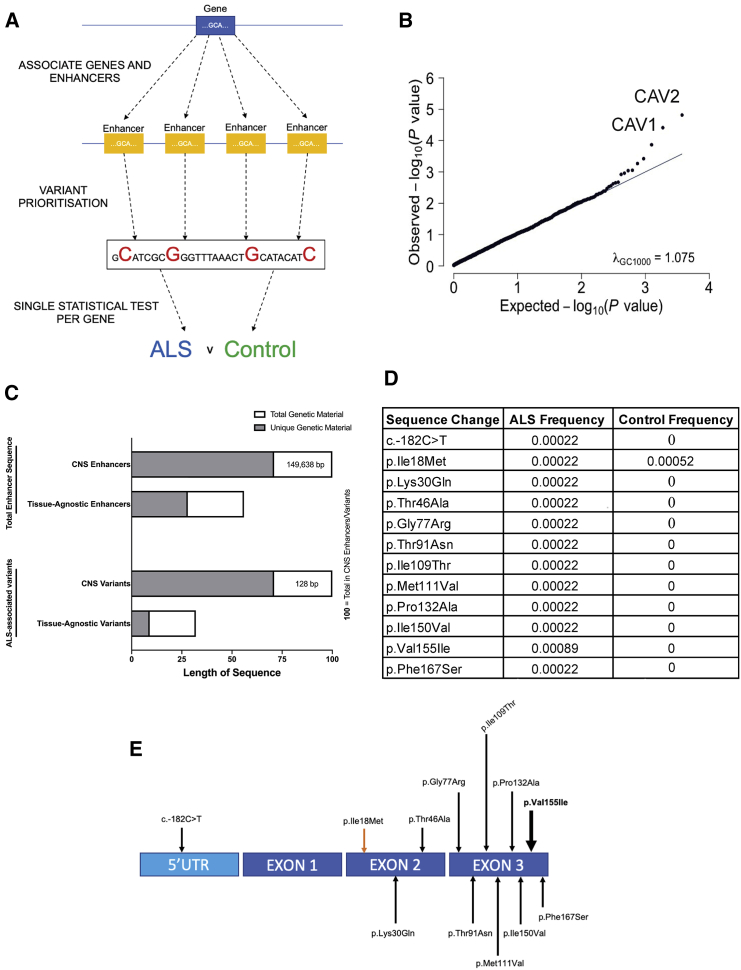
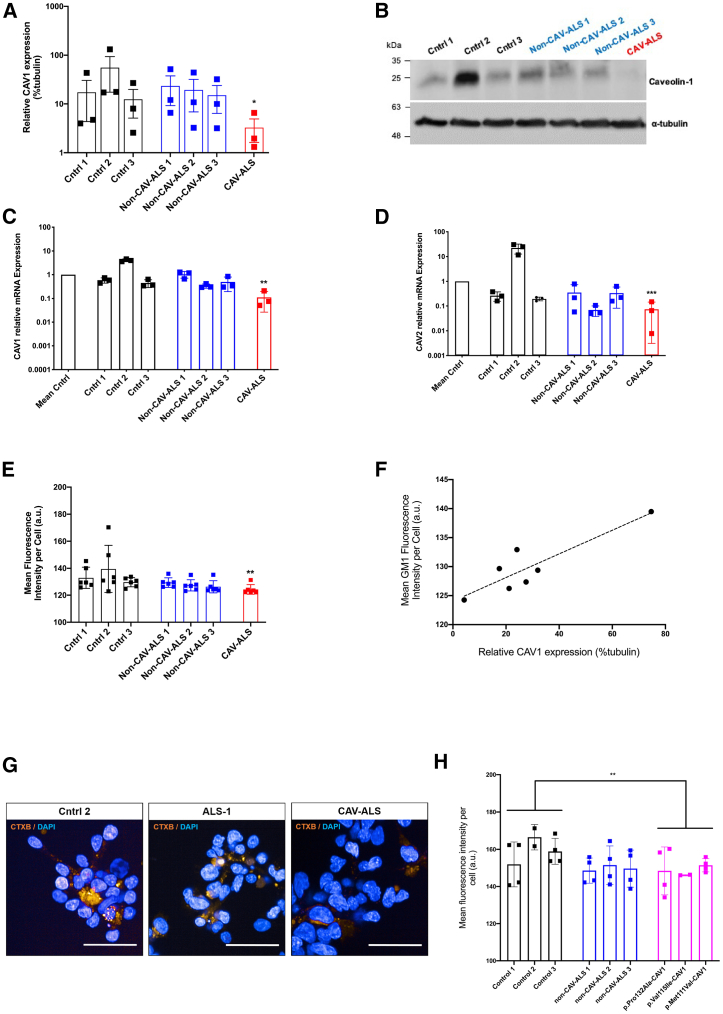
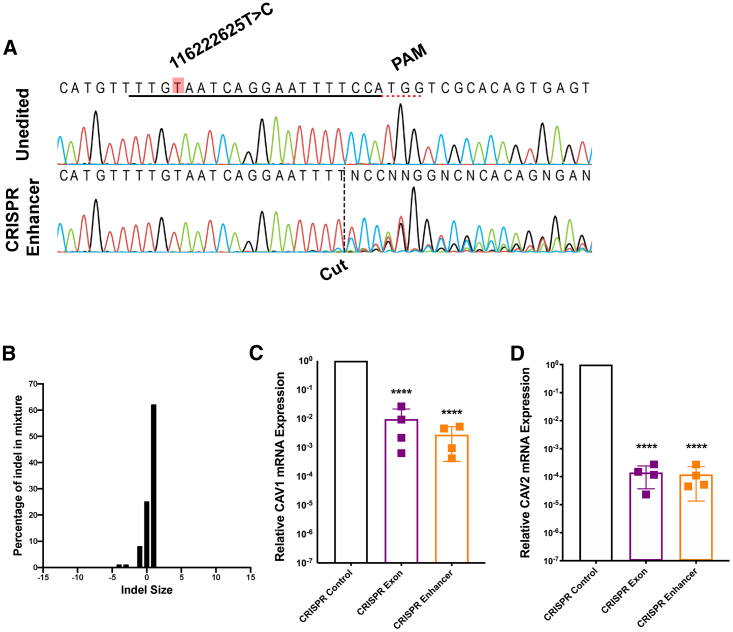
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disease. CAV1 and CAV2 organize membrane lipid rafts (MLRs) important for cell signaling and neuronal survival, and overexpression of CAV1 ameliorates ALS phenotypes in vivo. Genome-wide association studies localize a large proportion of ALS risk variants within the non-coding genome, but further characterization has been limited by lack of appropriate tools. By designing and applying a pipeline to identify pathogenic genetic variation within enhancer elements responsible for regulating gene expression, we identify disease-associated variation within CAV1/CAV2 enhancers, which replicate in an independent cohort. Discovered enhancer mutations reduce CAV1/CAV2 expression and disrupt MLRs in patient-derived cells, and CRISPR-Cas9 perturbation proximate to a patient mutation is sufficient to reduce CAV1/CAV2 expression in neurons. Additional enrichment of ALS-associated mutations within CAV1 exons positions CAV1 as an ALS risk gene. We propose CAV1/CAV2 overexpression as a personalized medicine target for ALS.
Keywords: CAV1; CAV2; amyotrophic lateral sclerosis; gene enhancers; membrane lipid rafts; non-coding DNA; whole-genome sequencing.
Copyright © 2020 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors declare no competing interests.
Figures
References
-
- Arganda-Carreras I., Fernández-González R., Muñoz-Barrutia A., Ortiz-De-Solorzano C. 3D reconstruction of histological sections: Application to mammary gland tissue. Microsc. Res. Tech. 2010;73:1019–1029. - PubMed
-
- Bemelmans A.-P., Husson I., Jaquet M., Mallet J., Kosofsky B.E., Gressens P. Lentiviral-mediated gene transfer of brain-derived neurotrophic factor is neuroprotective in a mouse model of neonatal excitotoxic challenge. J. Neurosci Res. 2006;83:50–60. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
