

Polymerization Reactions and Modifications of Polymers by Ionizing Radiation
- PMID: 33266261
- PMCID: PMC7760743
- DOI: 10.3390/polym12122877
Polymerization Reactions and Modifications of Polymers by Ionizing Radiation
Abstract
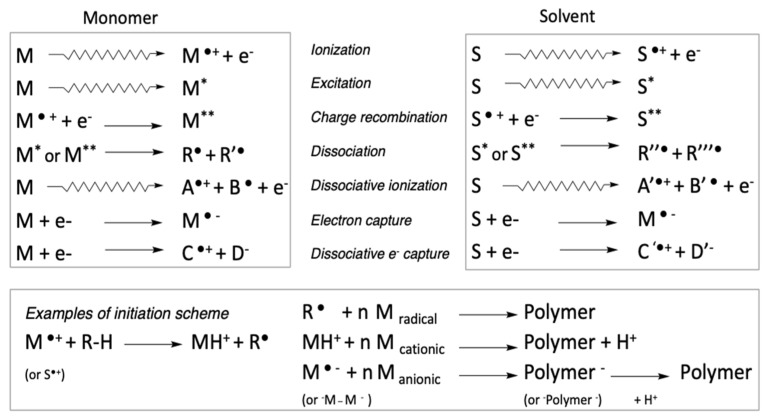
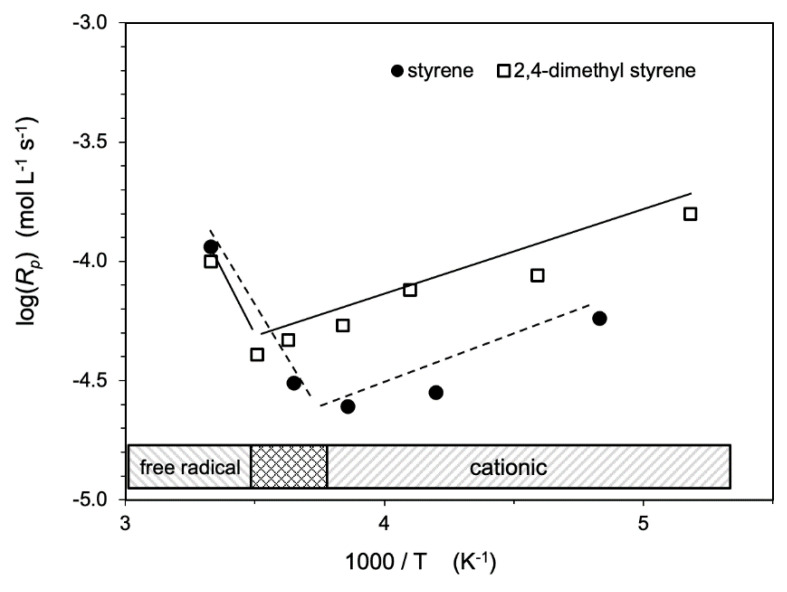
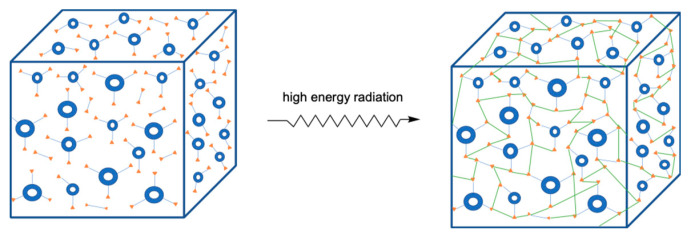
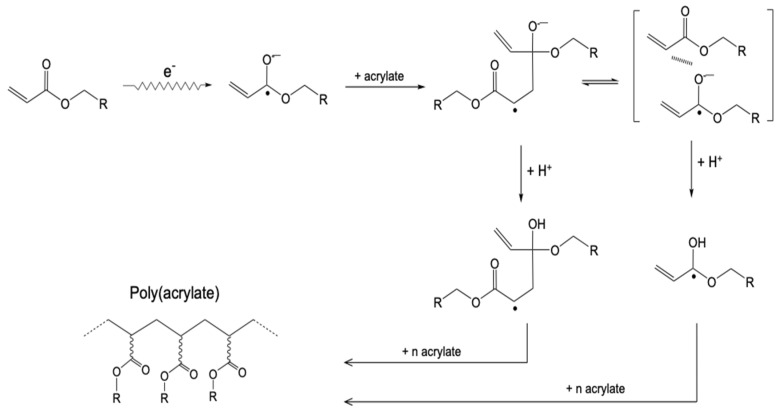
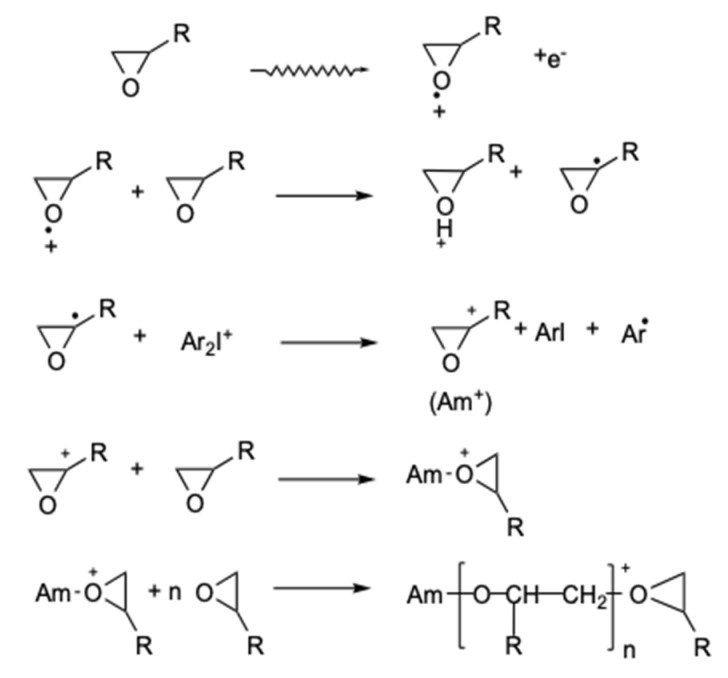
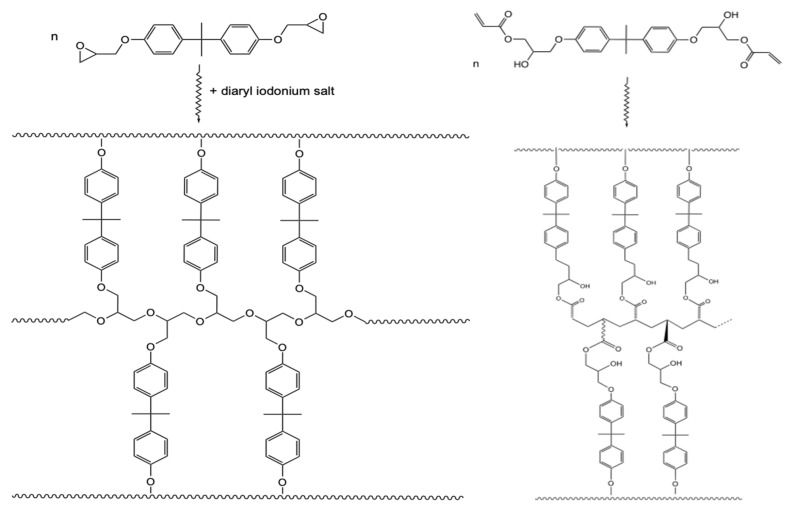
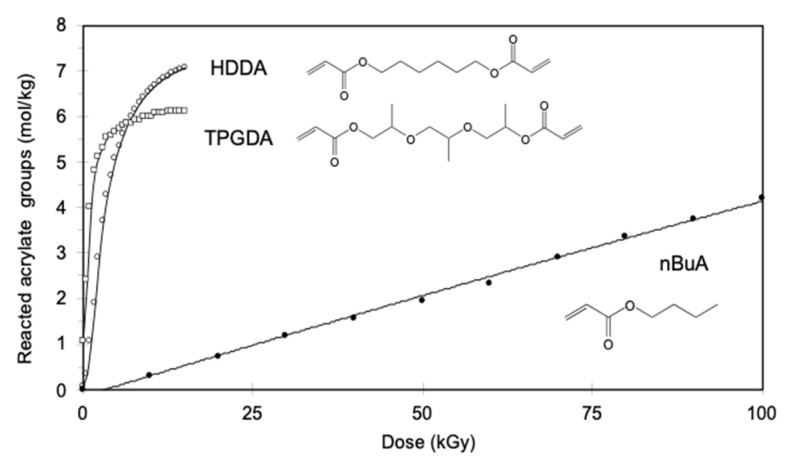
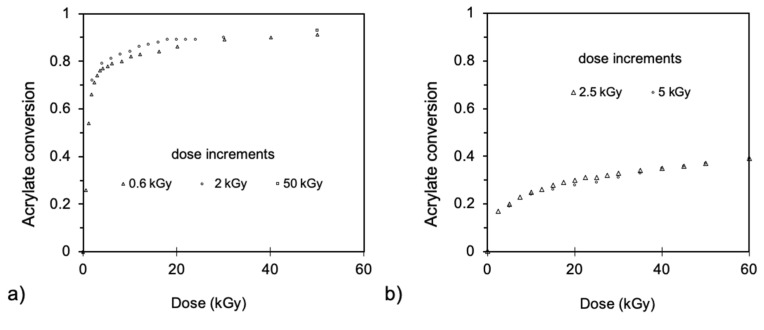
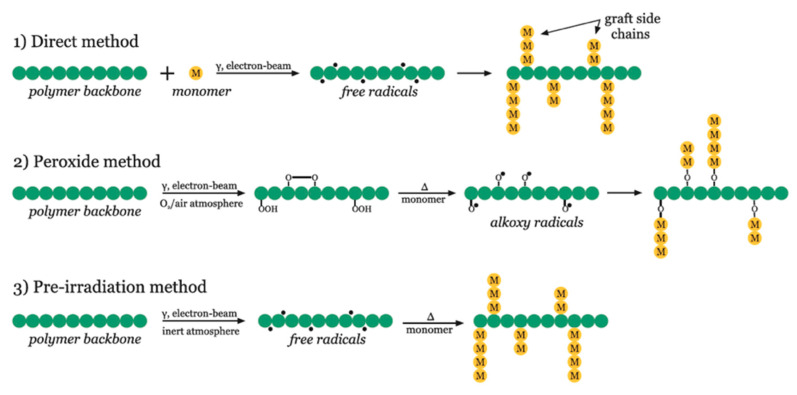
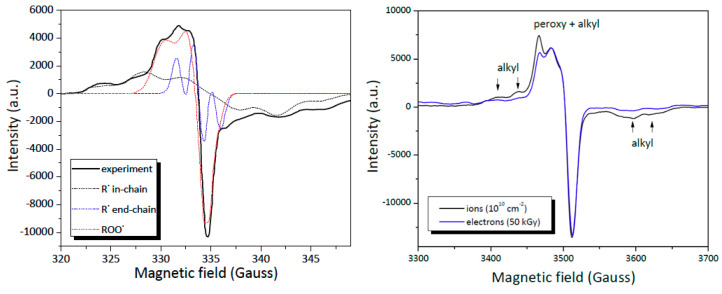
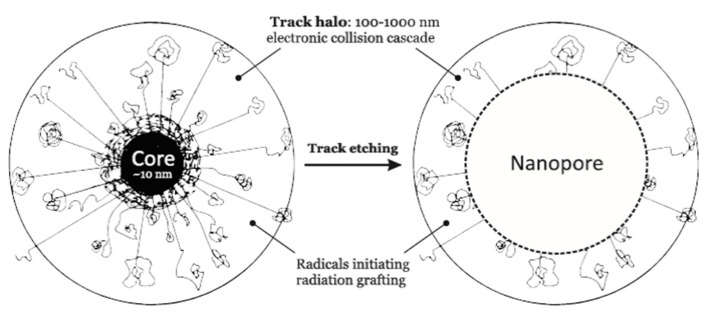
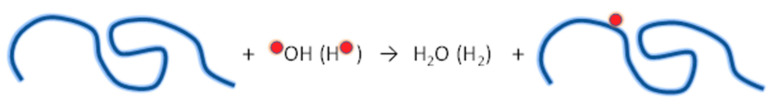
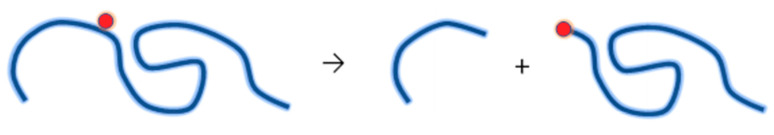
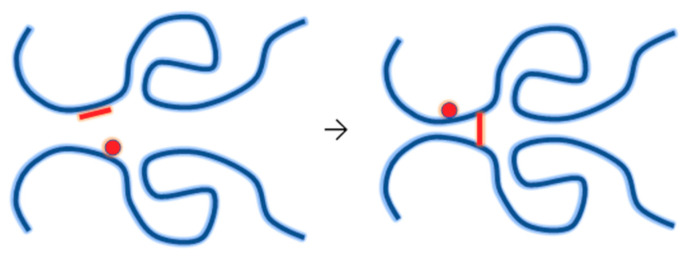
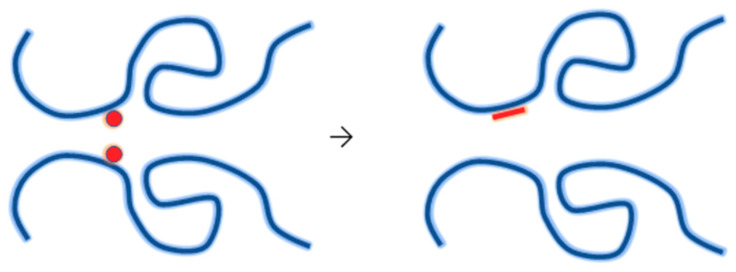
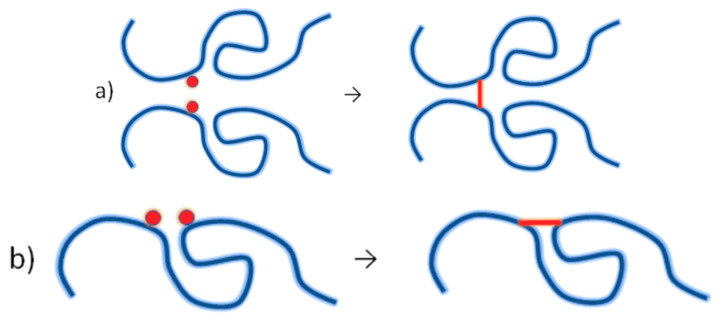
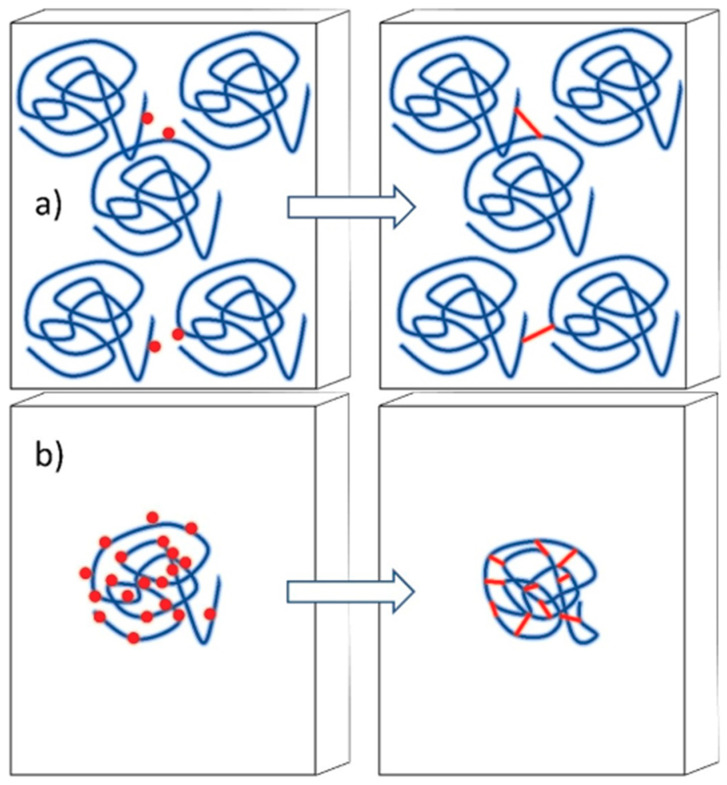
Ionizing radiation has become the most effective way to modify natural and synthetic polymers through crosslinking, degradation, and graft polymerization. This review will include an in-depth analysis of radiation chemistry mechanisms and the kinetics of the radiation-induced C-centered free radical, anion, and cation polymerization, and grafting. It also presents sections on radiation modifications of synthetic and natural polymers. For decades, low linear energy transfer (LLET) ionizing radiation, such as gamma rays, X-rays, and up to 10 MeV electron beams, has been the primary tool to produce many products through polymerization reactions. Photons and electrons interaction with polymers display various mechanisms. While the interactions of gamma ray and X-ray photons are mainly through the photoelectric effect, Compton scattering, and pair-production, the interactions of the high-energy electrons take place through coulombic interactions. Despite the type of radiation used on materials, photons or high energy electrons, in both cases ions and electrons are produced. The interactions between electrons and monomers takes place within less than a nanosecond. Depending on the dose rate (dose is defined as the absorbed radiation energy per unit mass), the kinetic chain length of the propagation can be controlled, hence allowing for some control over the degree of polymerization. When polymers are submitted to high-energy radiation in the bulk, contrasting behaviors are observed with a dominant effect of cross-linking or chain scission, depending on the chemical nature and physical characteristics of the material. Polymers in solution are subject to indirect effects resulting from the radiolysis of the medium. Likewise, for radiation-induced polymerization, depending on the dose rate, the free radicals generated on polymer chains can undergo various reactions, such as inter/intramolecular combination or inter/intramolecular disproportionation, b-scission. These reactions lead to structural or functional polymer modifications. In the presence of oxygen, playing on irradiation dose-rates, one can favor crosslinking reactions or promotes degradations through oxidations. The competition between the crosslinking reactions of C-centered free radicals and their reactions with oxygen is described through fundamental mechanism formalisms. The fundamentals of polymerization reactions are herein presented to meet industrial needs for various polymer materials produced or degraded by irradiation. Notably, the medical and industrial applications of polymers are endless and thus it is vital to investigate the effects of sterilization dose and dose rate on various polymers and copolymers with different molecular structures and morphologies. The presence or absence of various functional groups, degree of crystallinity, irradiation temperature, etc. all greatly affect the radiation chemistry of the irradiated polymers. Over the past decade, grafting new chemical functionalities on solid polymers by radiation-induced polymerization (also called RIG for Radiation-Induced Grafting) has been widely exploited to develop innovative materials in coherence with actual societal expectations. These novel materials respond not only to health emergencies but also to carbon-free energy needs (e.g., hydrogen fuel cells, piezoelectricity, etc.) and environmental concerns with the development of numerous specific adsorbents of chemical hazards and pollutants. The modification of polymers through RIG is durable as it covalently bonds the functional monomers. As radiation penetration depths can be varied, this technique can be used to modify polymer surface or bulk. The many parameters influencing RIG that control the yield of the grafting process are discussed in this review. These include monomer reactivity, irradiation dose, solvent, presence of inhibitor of homopolymerization, grafting temperature, etc. Today, the general knowledge of RIG can be applied to any solid polymer and may predict, to some extent, the grafting location. A special focus is on how ionizing radiation sources (ion and electron beams, UVs) may be chosen or mixed to combine both solid polymer nanostructuration and RIG. LLET ionizing radiation has also been extensively used to synthesize hydrogel and nanogel for drug delivery systems and other advanced applications. In particular, nanogels can either be produced by radiation-induced polymerization and simultaneous crosslinking of hydrophilic monomers in "nanocompartments", i.e., within the aqueous phase of inverse micelles, or by intramolecular crosslinking of suitable water-soluble polymers. The radiolytically produced oxidizing species from water, •OH radicals, can easily abstract H-atoms from the backbone of the dissolved polymers (or can add to the unsaturated bonds) leading to the formation of C-centered radicals. These C-centered free radicals can undergo two main competitive reactions; intramolecular and intermolecular crosslinking. When produced by electron beam irradiation, higher temperatures, dose rates within the pulse, and pulse repetition rates favour intramolecular crosslinking over intermolecular crosslinking, thus enabling a better control of particle size and size distribution. For other water-soluble biopolymers such as polysaccharides, proteins, DNA and RNA, the abstraction of H atoms or the addition to the unsaturation by •OH can lead to the direct scission of the backbone, double, or single strand breaks of these polymers.
Keywords: ionizing radiation; radiation induced grafting; radiation induced polymerization; radiation of natural polymers; radiation synthesis nanogels.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





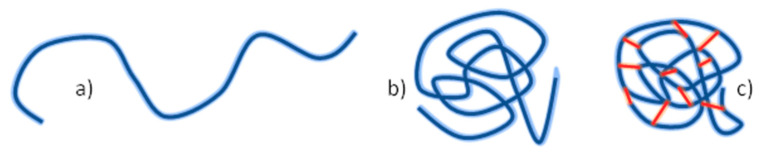
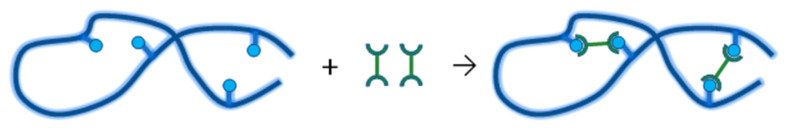

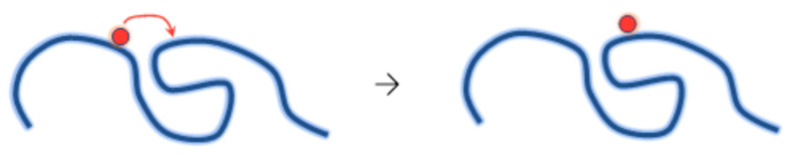
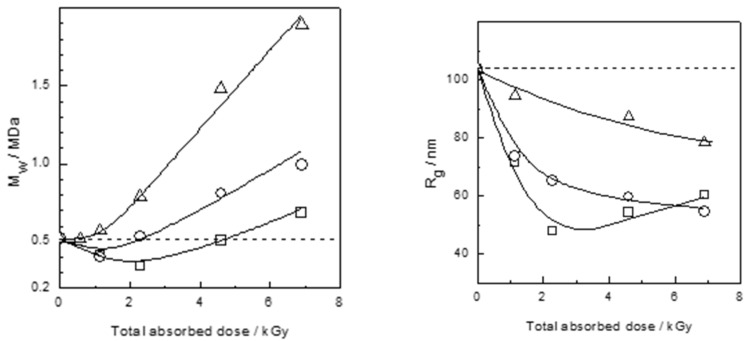

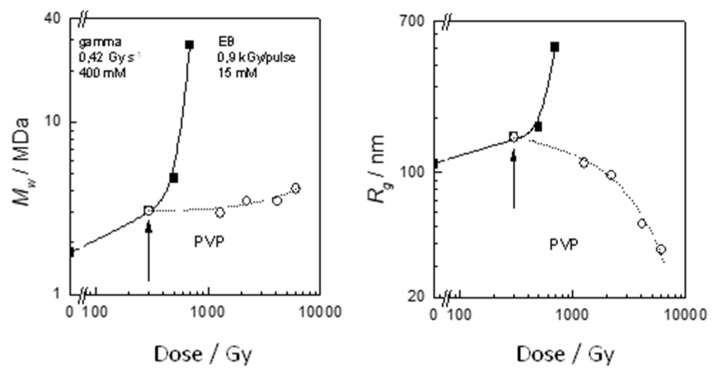
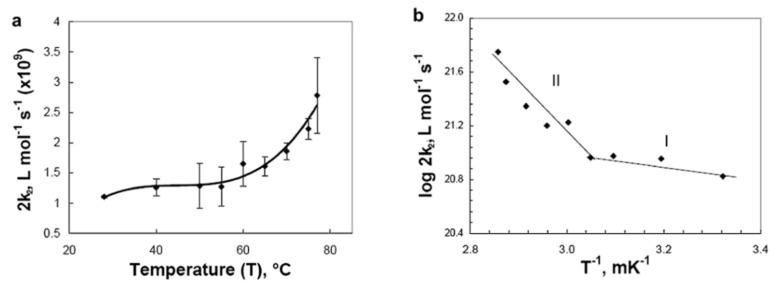

References
-
- Charlesby A. Atomic Radiation and Polymers. Pergamon Press; Oxford, UK: 1960.
-
- Chapiro A. Radiation Chemistry of Polymeric Systems. Interscience; New York, NY, USA: 1962.
-
- Dole M. The Radiation Chemistry of Macromolecules. Academic Press; New York, NY, USA: 1972.
-
- Schnabel W. Polymer Degradation. Principles and Practical Applications. Hanser; Muenchen, Germany: 1981.
-
- Rosiak J.M. Radiation Effects of Polymers. In: Clough R.L., Shalaby S.W., editors. ACS Symposium Series. Volume 475 American Chemical Society; Washington, DC, USA: 1991. ACS Symposium, Series.
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
