

A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies
- PMID: 33266957
- PMCID: PMC7514722
- DOI: 10.3390/e21030242
A Method Based on Differential Entropy-Like Function for Detecting Differentially Expressed Genes Across Multiple Conditions in RNA-Seq Studies
Abstract
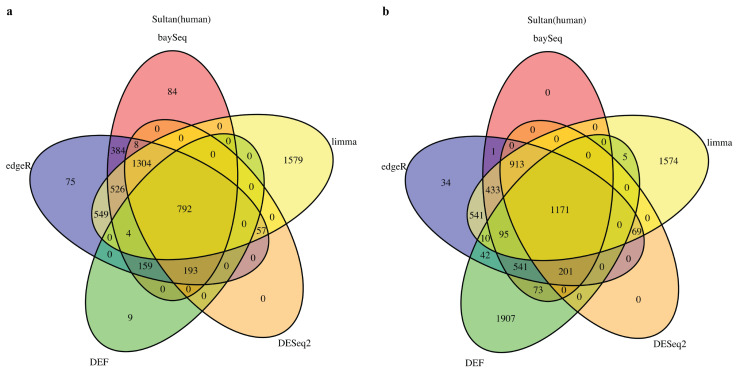
The advancement of high-throughput RNA sequencing has uncovered the profound truth in biology, ranging from the study of differential expressed genes to the identification of different genomic phenotype across multiple conditions. However, lack of biological replicates and low expressed data are still obstacles to measuring differentially expressed genes effectively. We present an algorithm based on differential entropy-like function (DEF) to test for the differential expression across time-course data or multi-sample data with few biological replicates. Compared with limma, edgeR, DESeq2, and baySeq, DEF maintains equivalent or better performance on the real data of two conditions. Moreover, DEF is well suited for predicting the genes that show the greatest differences across multiple conditions such as time-course data and identifies various biologically relevant genes.
Keywords: differential entropy-like function; differential expressed genes; multiple condition data; time-course data.
Conflict of interest statement
The authors declare no conflict of interest.
Figures





Similar articles
-
Robust identification of differentially expressed genes from RNA-seq data.Genomics. 2020 Mar;112(2):2000-2010. doi: 10.1016/j.ygeno.2019.11.012. Epub 2019 Nov 20. Genomics. 2020. PMID: 31756426
-
Three Differential Expression Analysis Methods for RNA Sequencing: limma, EdgeR, DESeq2.J Vis Exp. 2021 Sep 18;(175). doi: 10.3791/62528. J Vis Exp. 2021. PMID: 34605806
-
Getting the most out of RNA-seq data analysis.PeerJ. 2015 Oct 29;3:e1360. doi: 10.7717/peerj.1360. eCollection 2015. PeerJ. 2015. PMID: 26539333 Free PMC article.
-
A comparison of statistical methods for detecting differentially expressed genes from RNA-seq data.Am J Bot. 2012 Feb;99(2):248-56. doi: 10.3732/ajb.1100340. Epub 2012 Jan 20. Am J Bot. 2012. PMID: 22268221 Review.
-
RNA-Seq differential expression analysis: An extended review and a software tool.PLoS One. 2017 Dec 21;12(12):e0190152. doi: 10.1371/journal.pone.0190152. eCollection 2017. PLoS One. 2017. PMID: 29267363 Free PMC article. Review.
Cited by
-
Combining p-values from various statistical methods for microbiome data.Front Microbiol. 2022 Nov 10;13:990870. doi: 10.3389/fmicb.2022.990870. eCollection 2022. Front Microbiol. 2022. PMID: 36439799 Free PMC article.
-
Comparative study on differential expression analysis methods for single-cell RNA sequencing data with small biological replicates: Based on single-cell transcriptional data of PBMCs from COVID-19 severe patients.PLoS One. 2024 Mar 27;19(3):e0299358. doi: 10.1371/journal.pone.0299358. eCollection 2024. PLoS One. 2024. PMID: 38536877 Free PMC article.
References
-
- Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J., Salzberg S.L., Wold B.J., Pachter L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. - DOI - PMC - PubMed
-
- Clavijo B.J., Venturini L., Schudoma C., Accinelli G.G., Kaithakottil G., Wright J., Borrill P., Kettleborough G., Heavens D., Chapman H., et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017;27:885–896. doi: 10.1101/gr.217117.116. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
