

Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders
- PMID: 33268363
- PMCID: PMC7710361
- DOI: 10.1126/sciadv.aba8555
Landscapes of bacterial and metabolic signatures and their interaction in major depressive disorders
Abstract
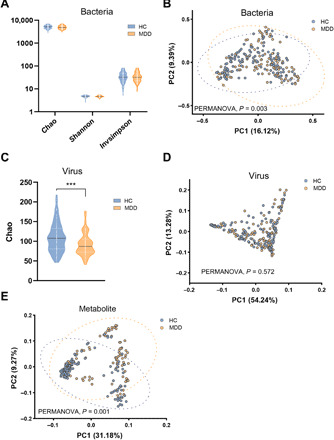
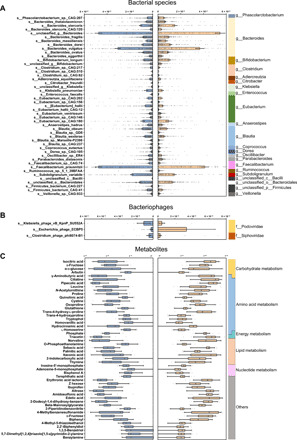
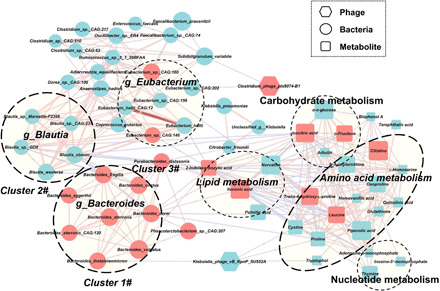
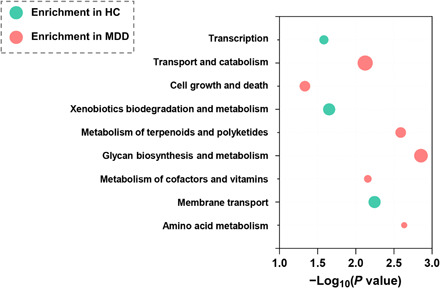
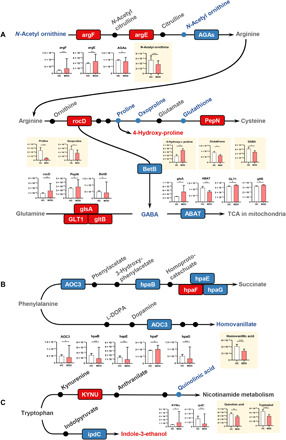
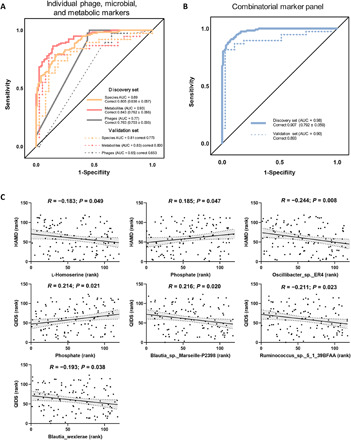
Gut microbiome disturbances have been implicated in major depressive disorder (MDD). However, little is known about how the gut virome, microbiome, and fecal metabolome change, and how they interact in MDD. Here, using whole-genome shotgun metagenomic and untargeted metabolomic methods, we identified 3 bacteriophages, 47 bacterial species, and 50 fecal metabolites showing notable differences in abundance between MDD patients and healthy controls (HCs). Patients with MDD were mainly characterized by increased abundance of the genus Bacteroides and decreased abundance of the genera Blautia and Eubacterium These multilevel omics alterations generated a characteristic MDD coexpression network. Disturbed microbial genes and fecal metabolites were consistently mapped to amino acid (γ-aminobutyrate, phenylalanine, and tryptophan) metabolism. Furthermore, we identified a combinatorial marker panel that robustly discriminated MDD from HC individuals in both the discovery and validation sets. Our findings provide a deep insight into understanding of the roles of disturbed gut ecosystem in MDD.
Copyright © 2020 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution NonCommercial License 4.0 (CC BY-NC).
Figures
References
-
- Kessler R. C., Berglund P., Demler O., Jin R., Koretz D., Merikangas K. R., Rush A. J., Walters E. E., Wang P. S., The epidemiology of major depressive disorder: Results from the National Comorbidity Survey Replication (NCS-R). JAMA 289, 3095–3105 (2003). - PubMed
-
- Mitchell A. J., Chan M., Bhatti H., Halton M., Grassi L., Johansen C., Meader N., Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: A meta-analysis of 94 interview-based studies. Lancet Oncol. 12, 160–174 (2011). - PubMed
-
- Fava M., Kendler K. S., Major depressive disorder. Neuron 28, 335–341 (2000). - PubMed
-
- Mitchell A. J., Vaze A., Rao S., Clinical diagnosis of depression in primary care: A meta-analysis. Lancet 374, 609–619 (2009). - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
