

Post-Translational Modifications of G Protein-Coupled Receptors Control Cellular Signaling Dynamics in Space and Time
- PMID: 33268549
- PMCID: PMC7736832
- DOI: 10.1124/pharmrev.120.000082
Post-Translational Modifications of G Protein-Coupled Receptors Control Cellular Signaling Dynamics in Space and Time
Abstract
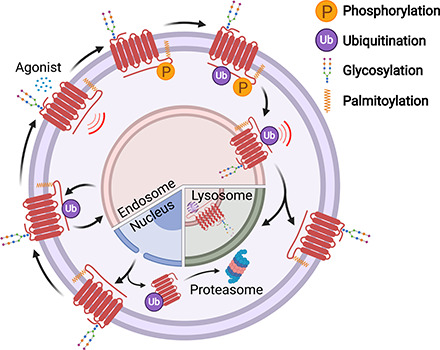
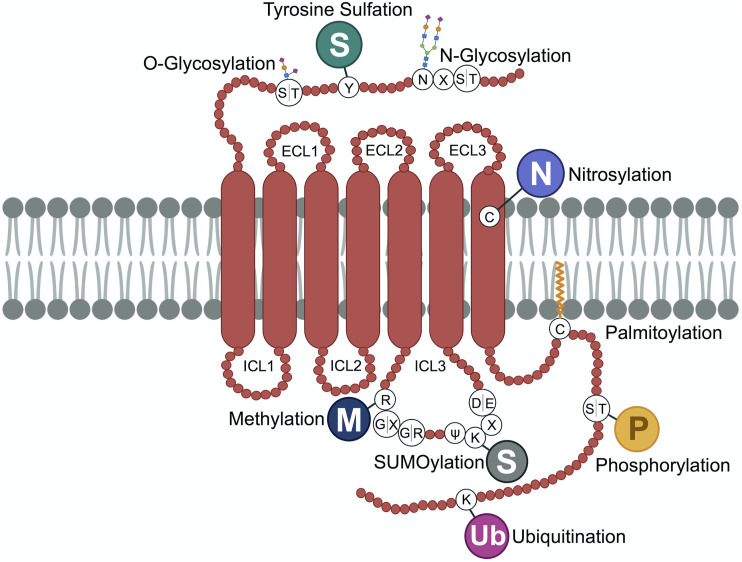
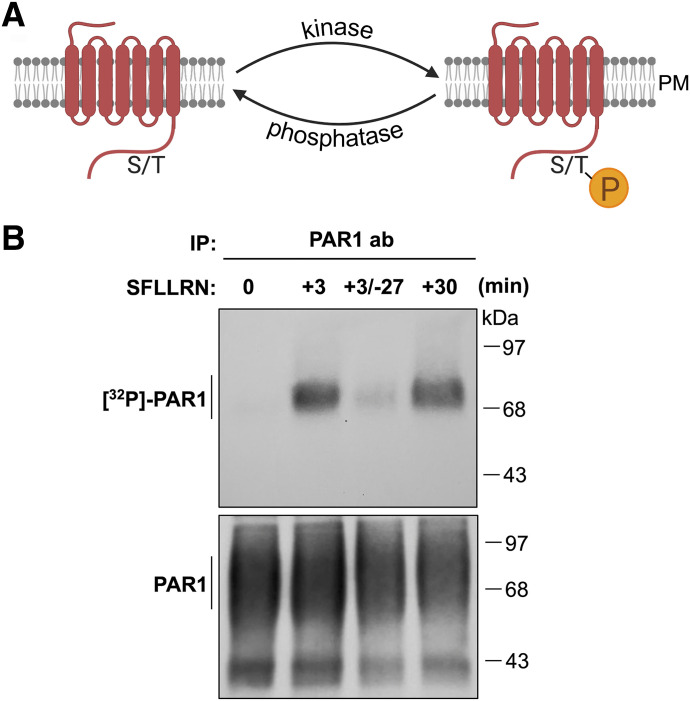
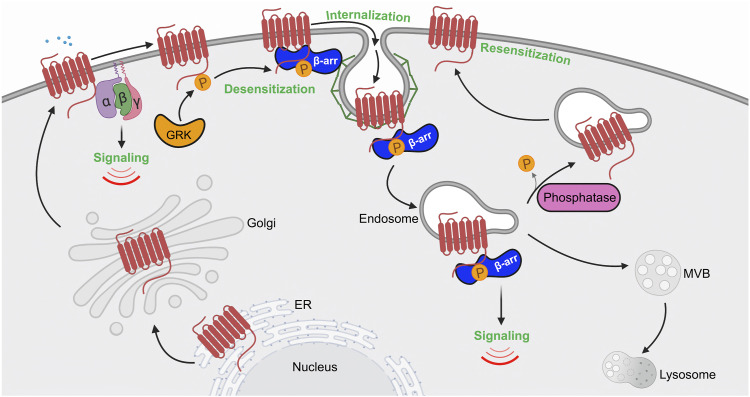
G protein-coupled receptors (GPCRs) are a large family comprising >800 signaling receptors that regulate numerous cellular and physiologic responses. GPCRs have been implicated in numerous diseases and represent the largest class of drug targets. Although advances in GPCR structure and pharmacology have improved drug discovery, the regulation of GPCR function by diverse post-translational modifications (PTMs) has received minimal attention. Over 200 PTMs are known to exist in mammalian cells, yet only a few have been reported for GPCRs. Early studies revealed phosphorylation as a major regulator of GPCR signaling, whereas later reports implicated a function for ubiquitination, glycosylation, and palmitoylation in GPCR biology. Although our knowledge of GPCR phosphorylation is extensive, our knowledge of the modifying enzymes, regulation, and function of other GPCR PTMs is limited. In this review we provide a comprehensive overview of GPCR post-translational modifications with a greater focus on new discoveries. We discuss the subcellular location and regulatory mechanisms that control post-translational modifications of GPCRs. The functional implications of newly discovered GPCR PTMs on receptor folding, biosynthesis, endocytic trafficking, dimerization, compartmentalized signaling, and biased signaling are also provided. Methods to detect and study GPCR PTMs as well as PTM crosstalk are further highlighted. Finally, we conclude with a discussion of the implications of GPCR PTMs in human disease and their importance for drug discovery. SIGNIFICANCE STATEMENT: Post-translational modification of G protein-coupled receptors (GPCRs) controls all aspects of receptor function; however, the detection and study of diverse types of GPCR modifications are limited. A thorough understanding of the role and mechanisms by which diverse post-translational modifications regulate GPCR signaling and trafficking is essential for understanding dysregulated mechanisms in disease and for improving and refining drug development for GPCRs.
Copyright © 2020 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
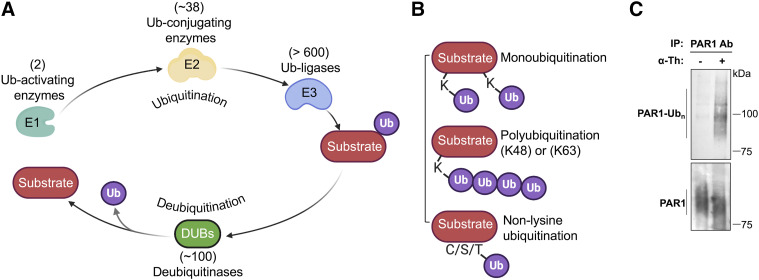
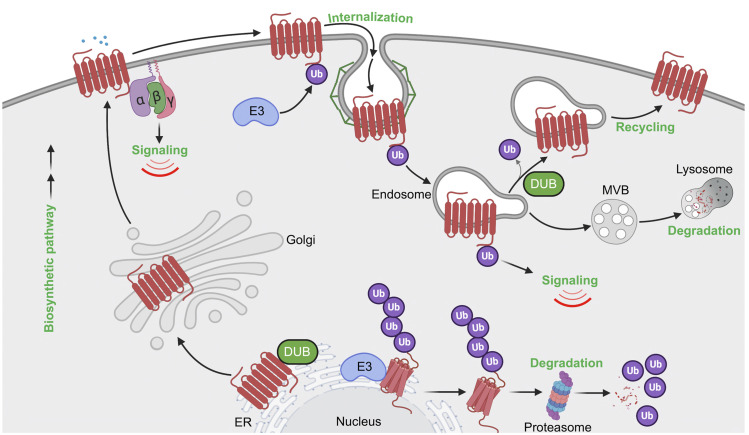
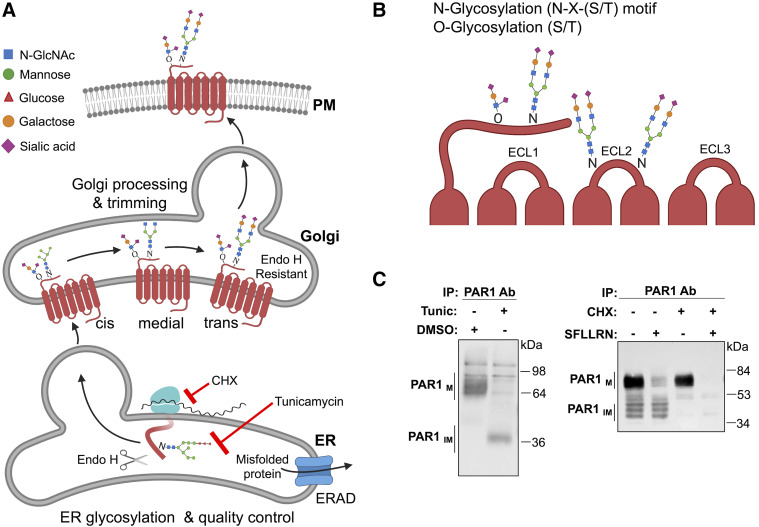
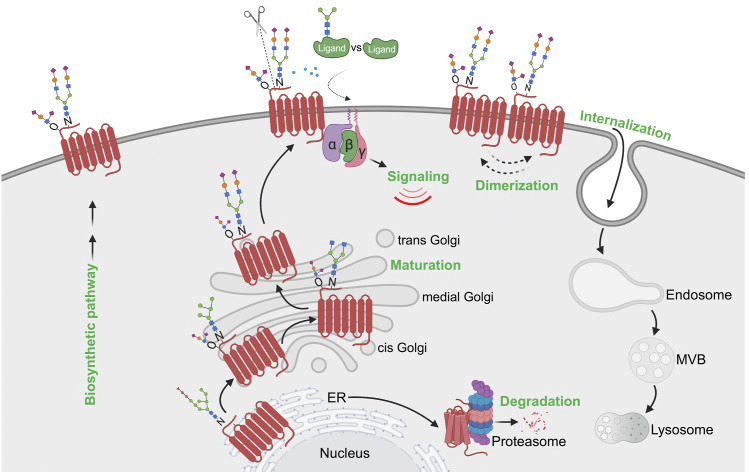
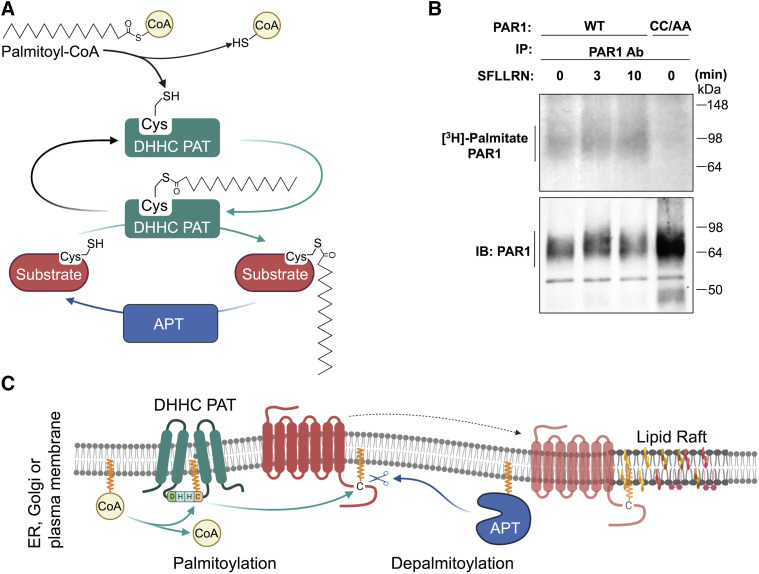
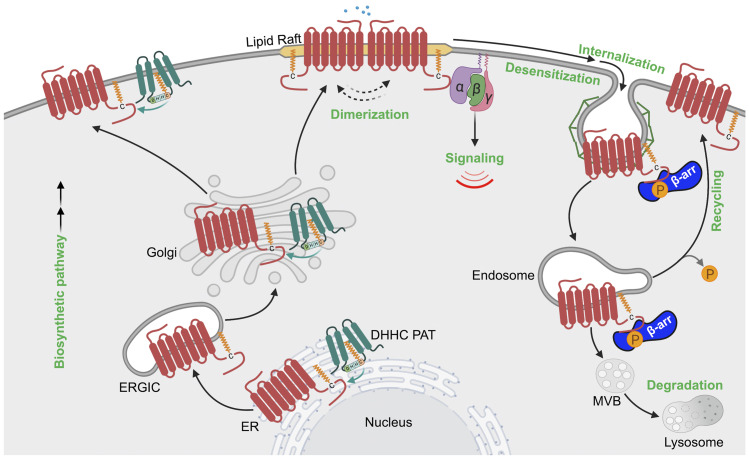
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
