

This is a preprint.
Fragment Binding to the Nsp3 Macrodomain of SARS-CoV-2 Identified Through Crystallographic Screening and Computational Docking
- PMID: 33269349
- PMCID: PMC7709169
- DOI: 10.1101/2020.11.24.393405
Fragment Binding to the Nsp3 Macrodomain of SARS-CoV-2 Identified Through Crystallographic Screening and Computational Docking
Update in
-
Fragment binding to the Nsp3 macrodomain of SARS-CoV-2 identified through crystallographic screening and computational docking.Sci Adv. 2021 Apr 14;7(16):eabf8711. doi: 10.1126/sciadv.abf8711. Print 2021 Apr. Sci Adv. 2021. PMID: 33853786 Free PMC article.
Abstract
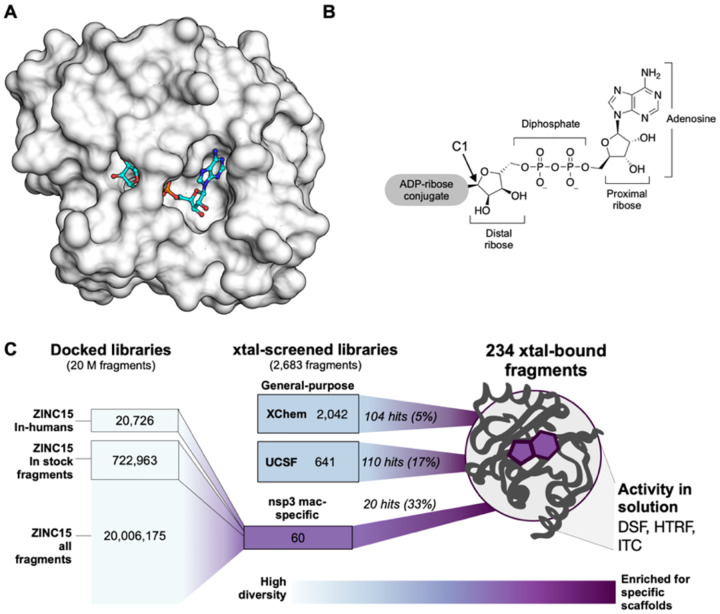
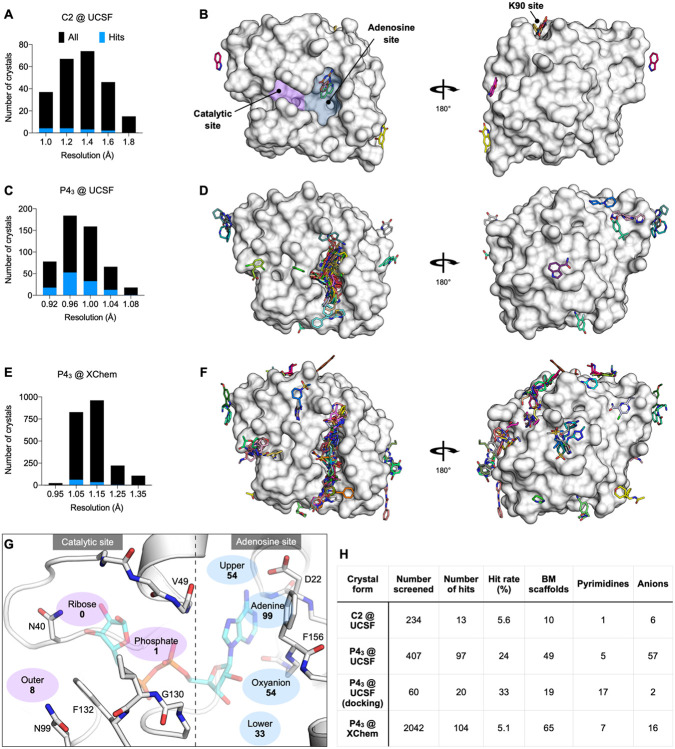
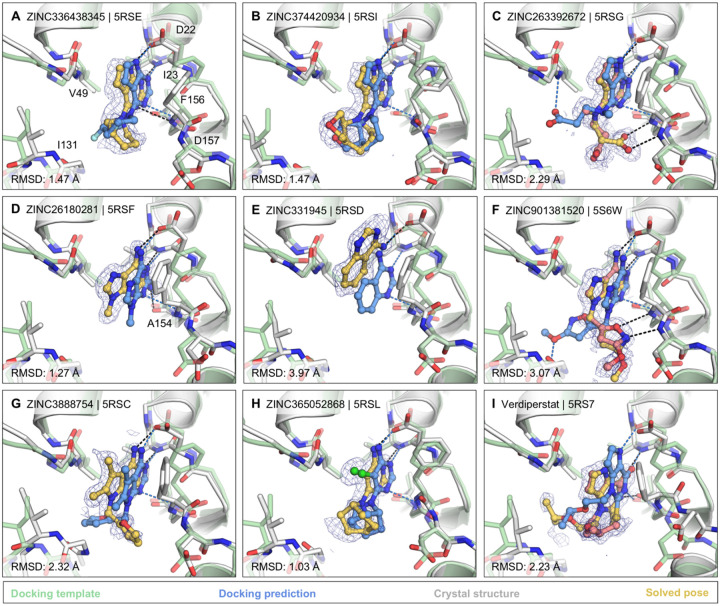
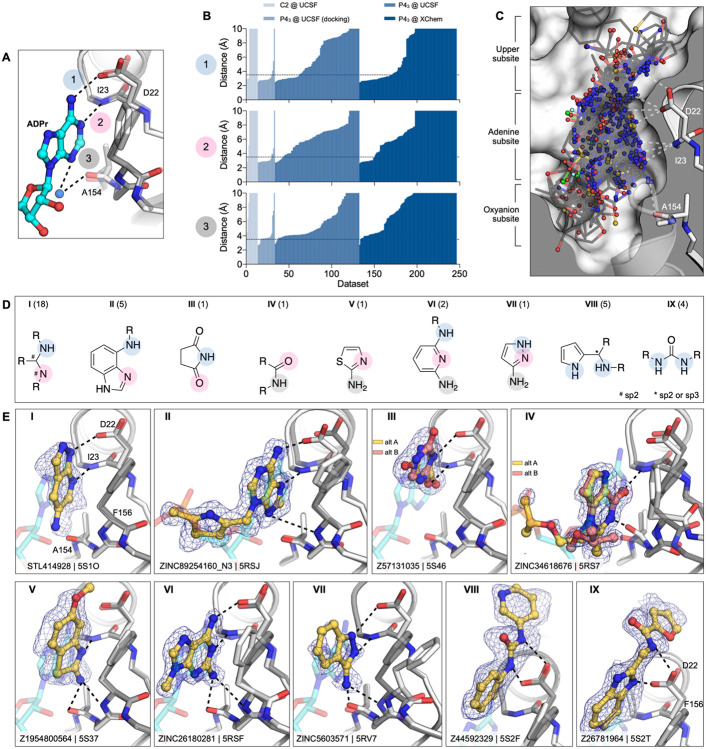
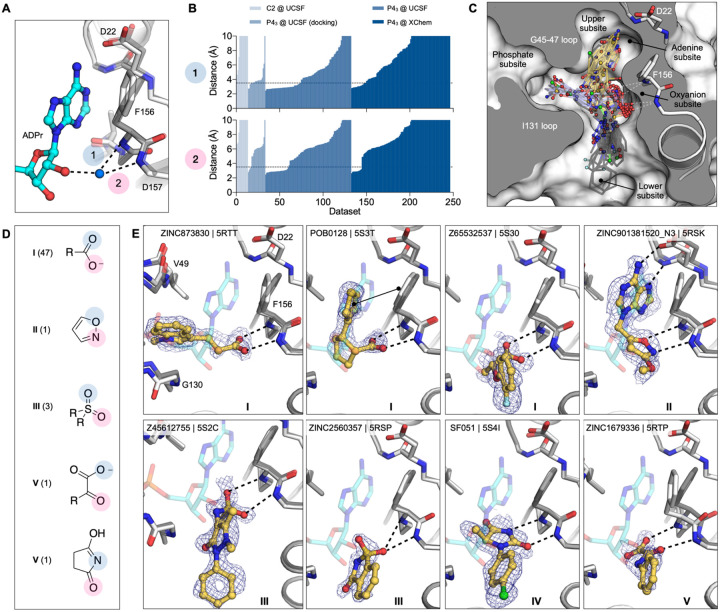
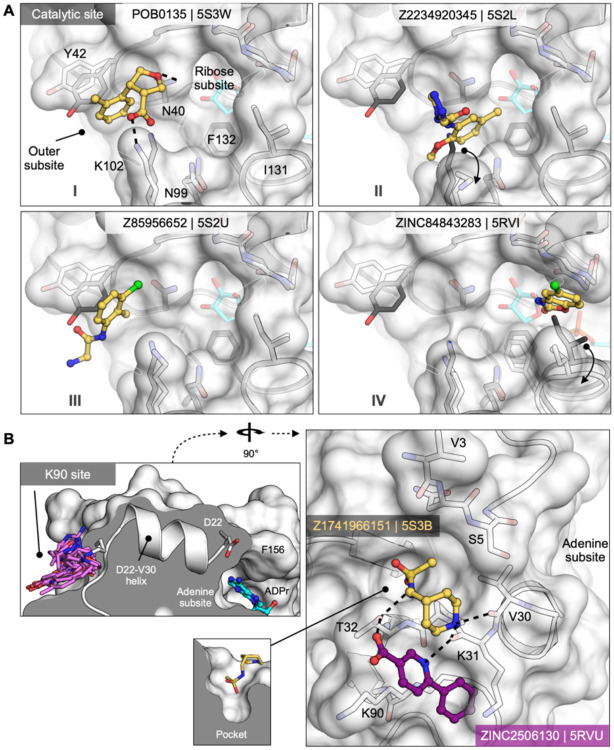
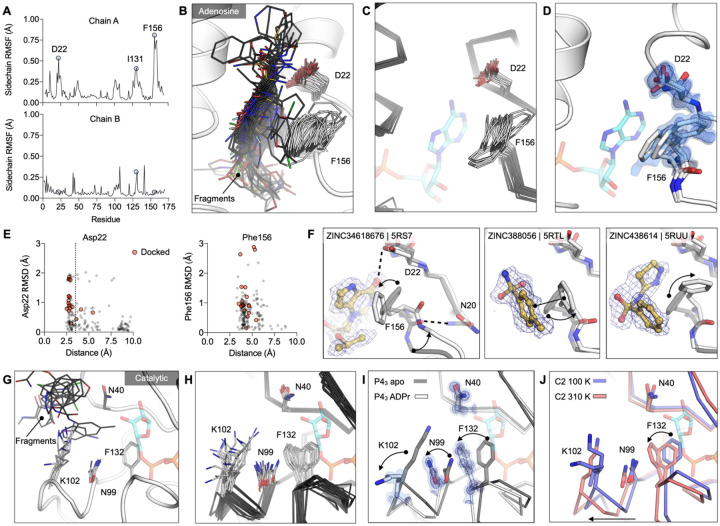
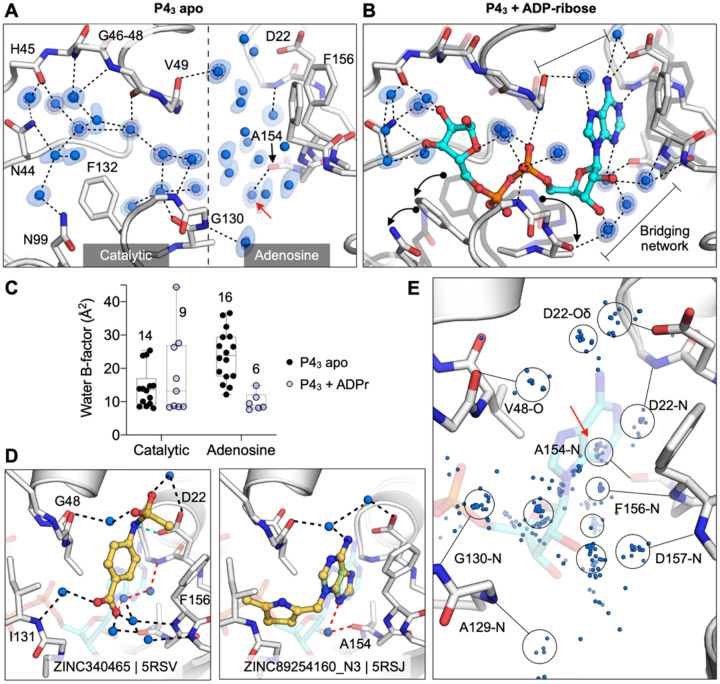
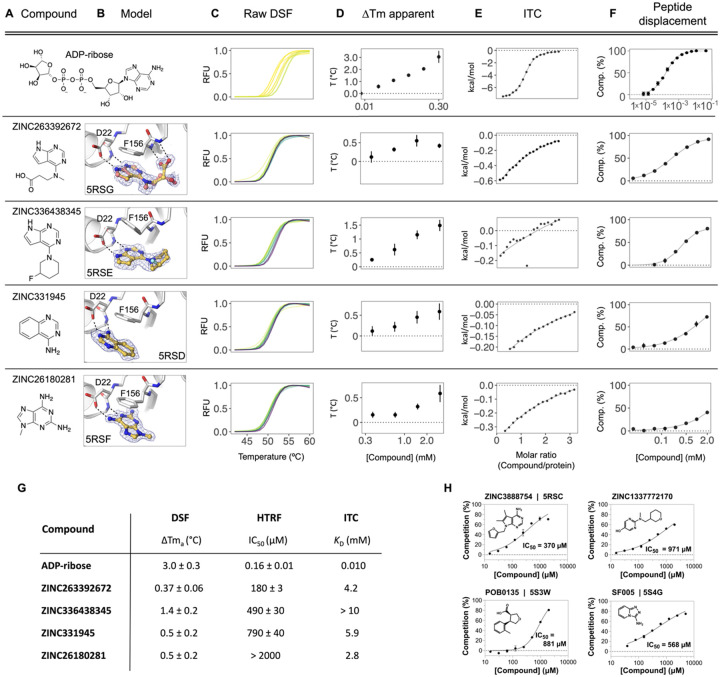
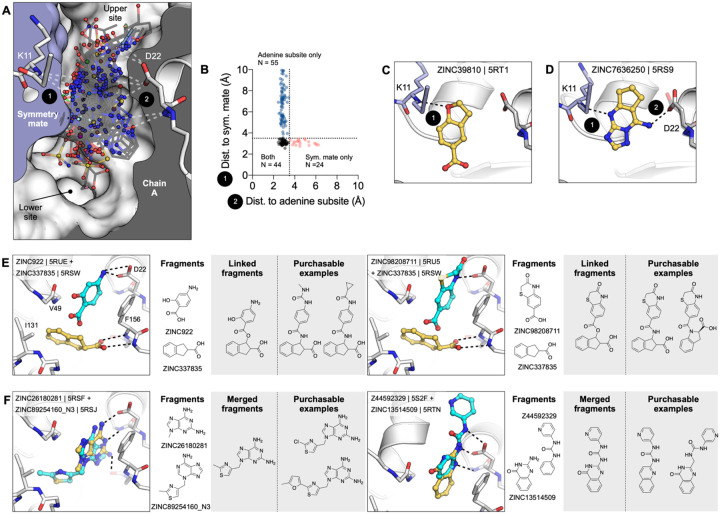
The SARS-CoV-2 macrodomain (Mac1) within the non-structural protein 3 (Nsp3) counteracts host-mediated antiviral ADP-ribosylation signalling. This enzyme is a promising antiviral target because catalytic mutations render viruses non-pathogenic. Here, we report a massive crystallographic screening and computational docking effort, identifying new chemical matter primarily targeting the active site of the macrodomain. Crystallographic screening of diverse fragment libraries resulted in 214 unique macrodomain-binding fragments, out of 2,683 screened. An additional 60 molecules were selected from docking over 20 million fragments, of which 20 were crystallographically confirmed. X-ray data collection to ultra-high resolution and at physiological temperature enabled assessment of the conformational heterogeneity around the active site. Several crystallographic and docking fragment hits were validated for solution binding using three biophysical techniques (DSF, HTRF, ITC). Overall, the 234 fragment structures presented explore a wide range of chemotypes and provide starting points for development of potent SARS-CoV-2 macrodomain inhibitors.
Conflict of interest statement
Competing interests
N. Jura is a member of the SAB of Turning Point Therapeutics and SUDO Biosciences. A. Ashworth is a co-founder of Tango Therapeutics, Azkarra Therapeutics, Ovibio Corporation; a consultant for SPARC, Bluestar, ProLynx, Earli, Cura, GenVivo and GSK; a member of the SAB of Genentech, GLAdiator, Circle and Cambridge Science Corporation; receives grant/research support from SPARC and AstraZeneca; holds patents on the use of PARP inhibitors held jointly with AstraZeneca which he has benefitted financially (and may do so in the future). B. Shoichet and J. Irwin are co-founders of a company, BlueDolphin LLC, that does fee-for-service docking. J. Fraser is a founder of Keyhole Therapeutics and a shareholder of Relay Therapeutics and Keyhole Therapeutics. The Fraser laboratory has received sponsored research support from Relay Therapeutics.
Figures
References
-
- Rack J. G. M., Perina D., Ahel I., Macrodomains: Structure, Function, Evolution, and Catalytic Activities. Annu. Rev. Biochem. 85, 431–454 (2016). - PubMed