

Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention
- PMID: 33274002
- PMCID: PMC7683149
- DOI: 10.1155/2020/5021694
Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention
Abstract
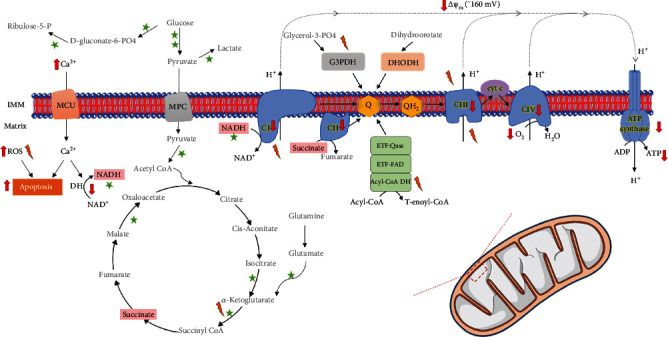
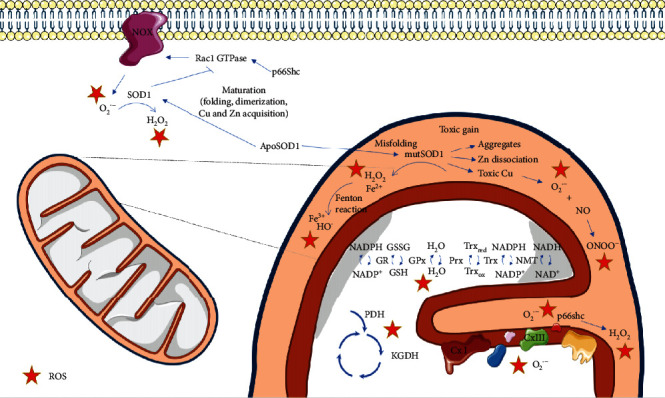
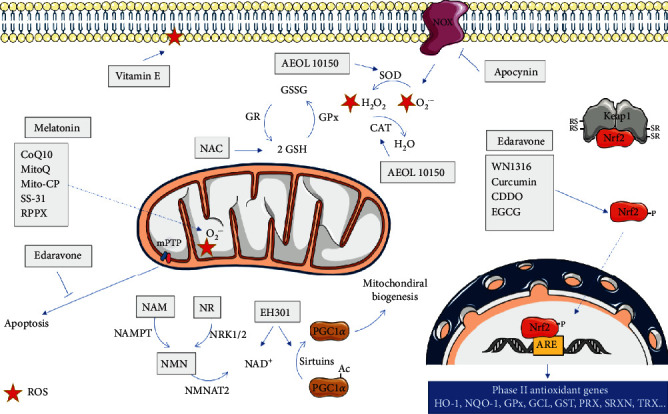
Amyotrophic lateral sclerosis (ALS), also known as Lou Gehrig's disease or Charcot disease, is a fatal neurodegenerative disease that affects motor neurons (MNs) and leads to death within 2-5 years of diagnosis, without any effective therapy available. Although the pathological mechanisms leading to ALS are still unknown, a wealth of evidence indicates that an excessive reactive oxygen species (ROS) production associated with an inefficient antioxidant defense represents an important pathological feature in ALS. Substantial evidence indicates that oxidative stress (OS) is implicated in the loss of MNs and in mitochondrial dysfunction, contributing decisively to neurodegeneration in ALS. Although the modulation of OS represents a promising approach to protect MNs from degeneration, the fact that several antioxidants with beneficial effects in animal models failed to show any therapeutic benefit in patients raises several questions that should be analyzed. Using specific queries for literature search on PubMed, we review here the role of OS-related mechanisms in ALS, including the involvement of altered mitochondrial function with repercussions in neurodegeneration. We also describe antioxidant compounds that have been mostly tested in preclinical and clinical trials of ALS, also describing their respective mechanisms of action. While the description of OS mechanism in the different mutations identified in ALS has as principal objective to clarify the contribution of OS in ALS, the description of positive and negative outcomes for each antioxidant is aimed at paving the way for novel opportunities for intervention. In conclusion, although antioxidant strategies represent a very promising approach to slow the progression of the disease, it is of utmost need to invest on the characterization of OS profiles representative of each subtype of patient, in order to develop personalized therapies, allowing to understand the characteristics of antioxidants that have beneficial effects on different subtypes of patients.
Copyright © 2020 Teresa Cunha-Oliveira et al.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- Tanaka M., Sakata T., Palumbo J., Akimoto M. A 24-week, phase III, double-blind, parallel-group study of edaravone (MCI-186) for treatment of amyotrophic lateral sclerosis (ALS) (P3.189) Neurology. 2016;86
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
