

Mitochondrial oxidative function in NAFLD: Friend or foe?
- PMID: 33276146
- PMCID: PMC8324685
- DOI: 10.1016/j.molmet.2020.101134
Mitochondrial oxidative function in NAFLD: Friend or foe?
Abstract
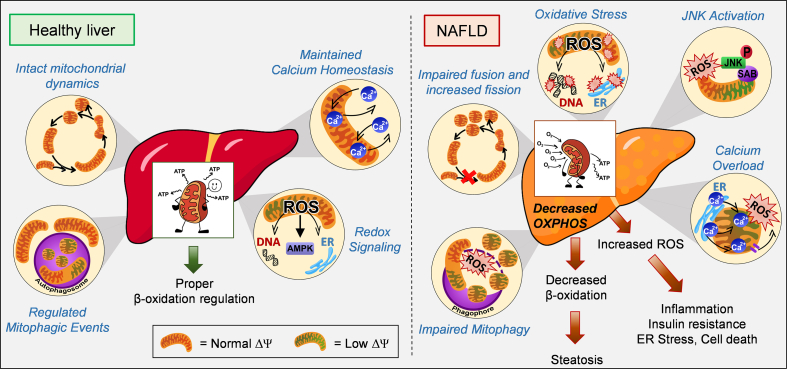
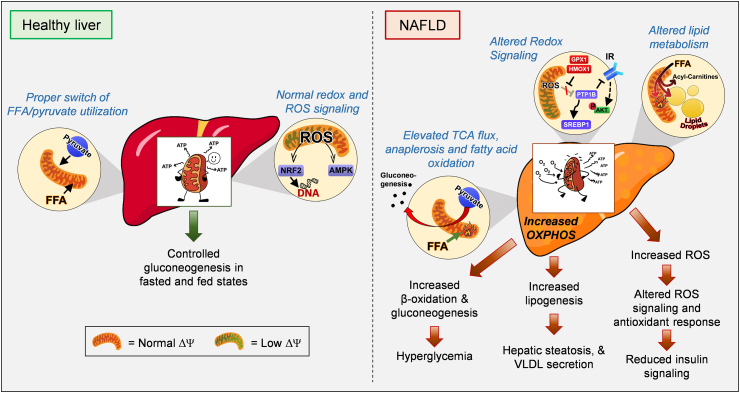
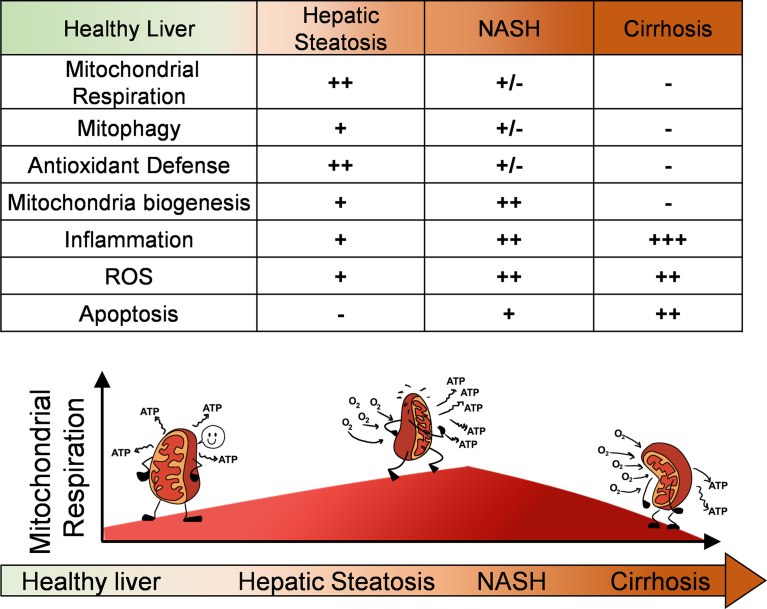
Background: Mitochondrial oxidative function plays a key role in the development of non-alcoholic fatty liver disease (NAFLD) and insulin resistance (IR). Recent studies reported that fatty liver might not be a result of decreased mitochondrial fat oxidation caused by mitochondrial damage. Rather, NAFLD and IR induce an elevation in mitochondrial function that covers the increased demand for carbon intermediates and ATP caused by elevated lipogenesis and gluconeogenesis. Furthermore, mitochondria play a role in regulating hepatic insulin sensitivity and lipogenesis by modulating redox-sensitive signaling pathways.
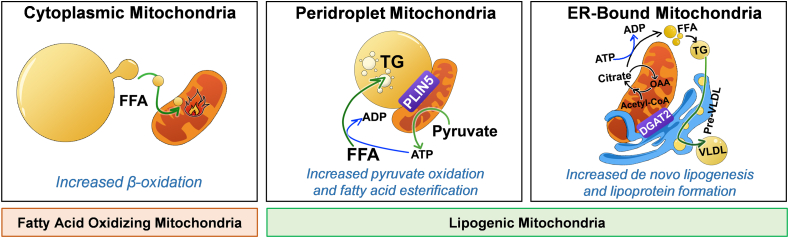
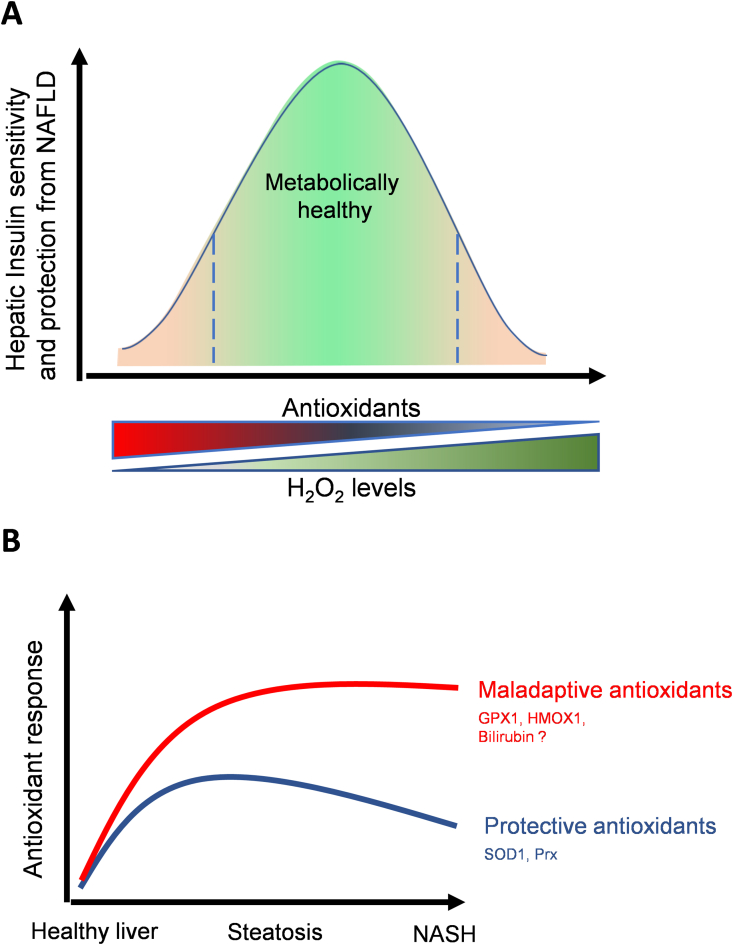
Scope of review: We review the contradictory studies indicating that NAFLD and hyperglycemia can either increase or decrease mitochondrial oxidative capacity in the liver. We summarize mechanisms regulating mitochondrial heterogeneity inside the same cell and discuss how these mechanisms may determine the role of mitochondria in NAFLD. We further discuss the role of endogenous antioxidants in controlling mitochondrial H2O2 release and redox-mediated signaling. We describe the emerging concept that the subcellular location of cellular antioxidants is a key determinant of their effects on NAFLD.
Major conclusions: The balance of fat oxidation versus accumulation depends on mitochondrial fuel preference rather than ATP-synthesizing respiration. As such, therapies targeting fuel preference might be more suitable for treating NAFLD. Similarly, suppressing maladaptive antioxidants, rather than interfering with physiological mitochondrial H2O2-mediated signaling, may allow the maintenance of intact hepatic insulin signaling in NAFLD. Exploration of the subcellular compartmentalization of different antioxidant systems and the unique functions of specific mitochondrial subpopulations may offer new intervention points to treat NAFLD.
Keywords: H(2)O(2); Lipid metabolism; Mitochondria; Mitochondrial heterogeneity; Mitophagy; NAFLD; NASH.
Copyright © 2020 The Author(s). Published by Elsevier GmbH.. All rights reserved.
Figures
References
-
- Rolfe D.F., Brown G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiological Reviews. 1997;77(3):731–758. - PubMed
-
- McGarry J.D., Foster D.W. Effects of exogenous fatty acid concentration on glucagon-induced changes in hepatic fatty acid metabolism. Diabetes. 1980;29(3):236–240. - PubMed
-
- Bril F., Lomonaco R., Orsak B., Ortiz-Lopez C., Webb A., Tio F. Relationship between disease severity, hyperinsulinemia, and impaired insulin clearance in patients with nonalcoholic steatohepatitis. Hepatology. 2014;59(6):2178–2187. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
