

Genetic and epigenetic determinants of diffuse large B-cell lymphoma
- PMID: 33277464
- PMCID: PMC7718920
- DOI: 10.1038/s41408-020-00389-w
Genetic and epigenetic determinants of diffuse large B-cell lymphoma
Abstract
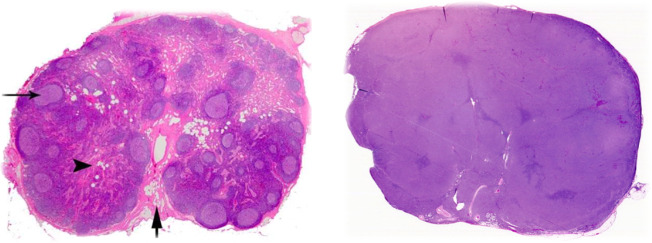
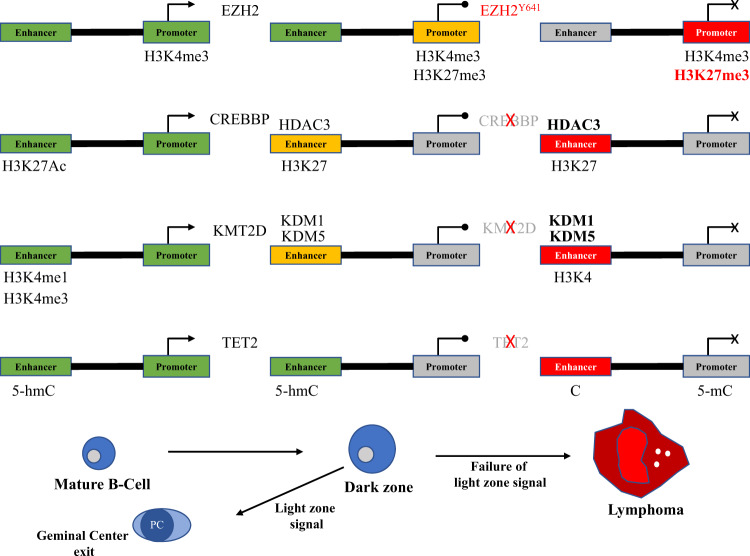
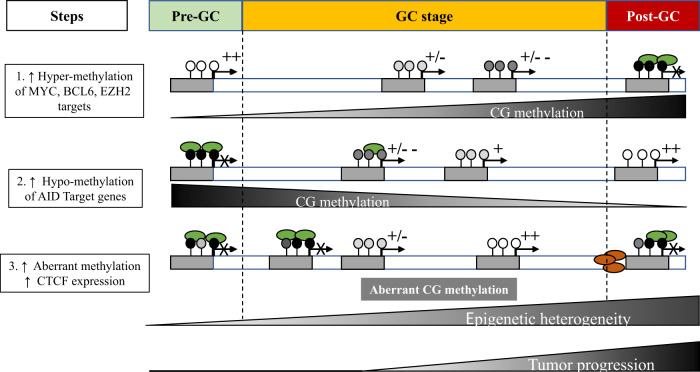
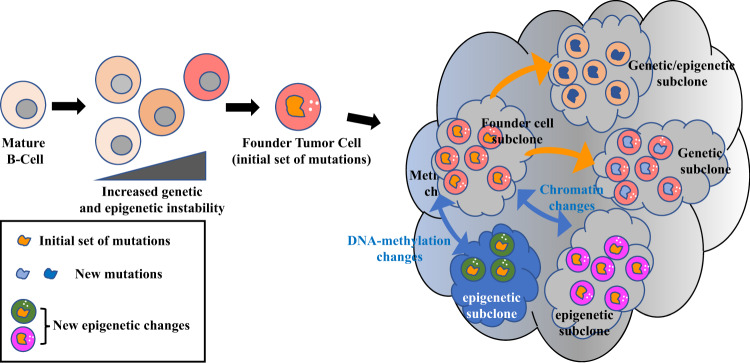
Diffuse large B-cell lymphoma (DLBCL) is the most common type of lymphoma and is notorious for its heterogeneity, aggressive nature, and the frequent development of resistance and/or relapse after treatment with standard chemotherapy. To address these problems, a strong emphasis has been placed on researching the molecular origins and mechanisms of DLBCL to develop effective treatments. One of the major insights produced by such research is that DLBCL almost always stems from genetic damage that occurs during the germinal center (GC) reaction, which is required for the production of high-affinity antibodies. Indeed, there is significant overlap between the mechanisms that govern the GC reaction and those that drive the progression of DLBCL. A second important insight is that some of the most frequent genetic mutations that occur in DLBCL are those related to chromatin and epigenetics, especially those related to proteins that "write" histone post-translational modifications (PTMs). Mutation or deletion of these epigenetic writers often renders cells unable to epigenetically "switch on" critical gene sets that are required to exit the GC reaction, differentiate, repair DNA, and other essential cellular functions. Failure to activate these genes locks cells into a genotoxic state that is conducive to oncogenesis and/or relapse.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
-
- Swerdlow S. H., International Agency for Research on Cancer & World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th edn, 439 p. (International Agency for Research on Cancer, Lyon, France, 2008).
-
- Swerdlow S. H., World Health Organization & International Agency for Research on Cancer. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, revised 4th edn, 585 p. (International Agency for Research on Cancer, Lyon, France, 2017).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
