

Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5)
- PMID: 33280500
- PMCID: PMC8526012
- DOI: 10.1080/15548627.2020.1851897
Autophagy alleviates hypoxia-induced blood-brain barrier injury via regulation of CLDN5 (claudin 5)
Abstract
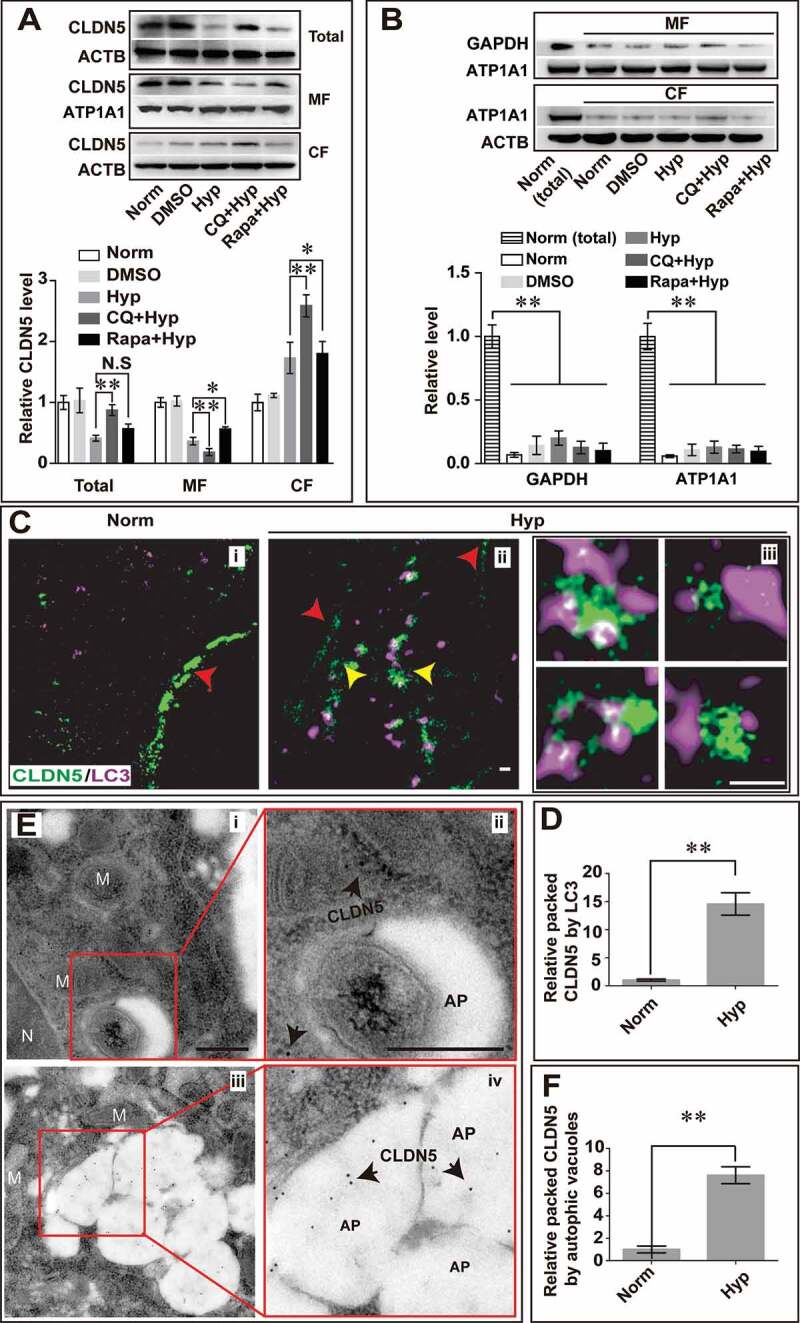
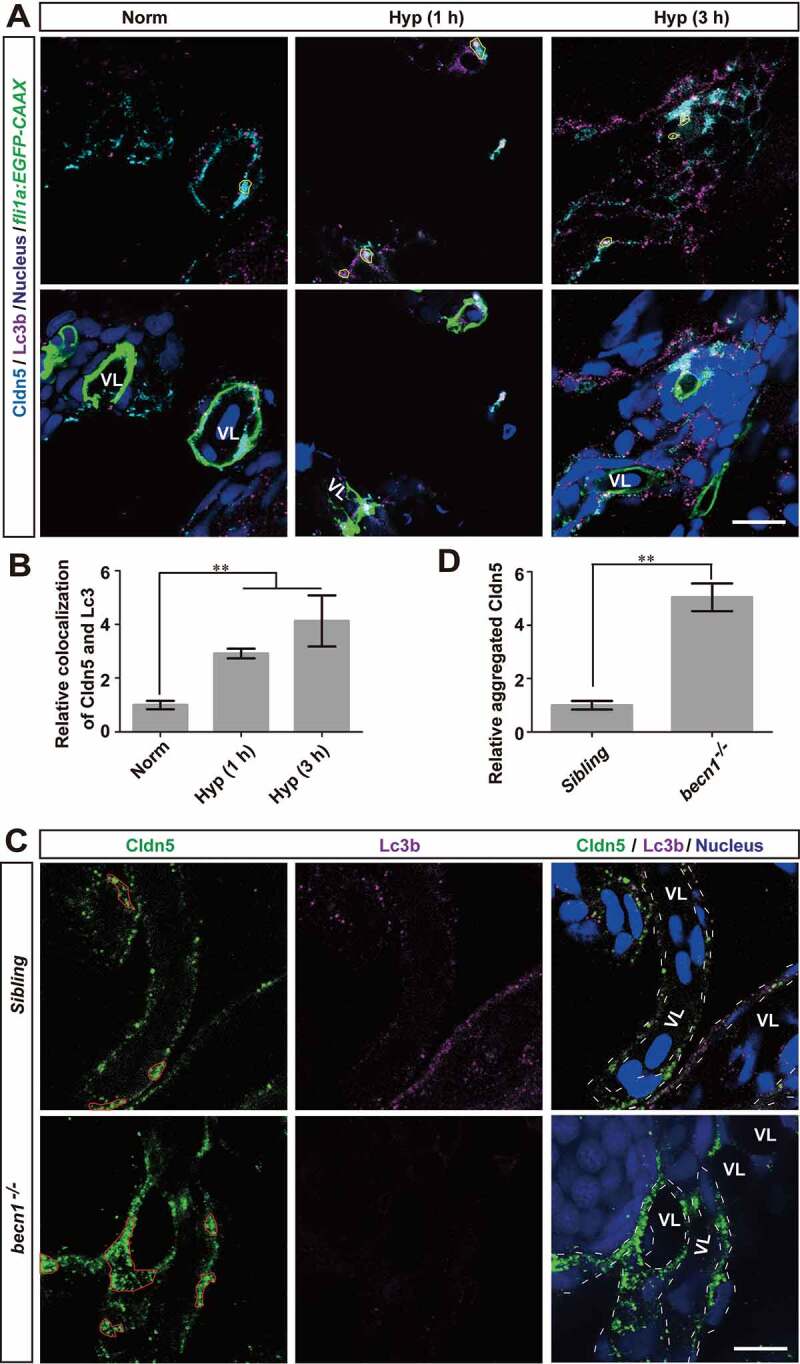
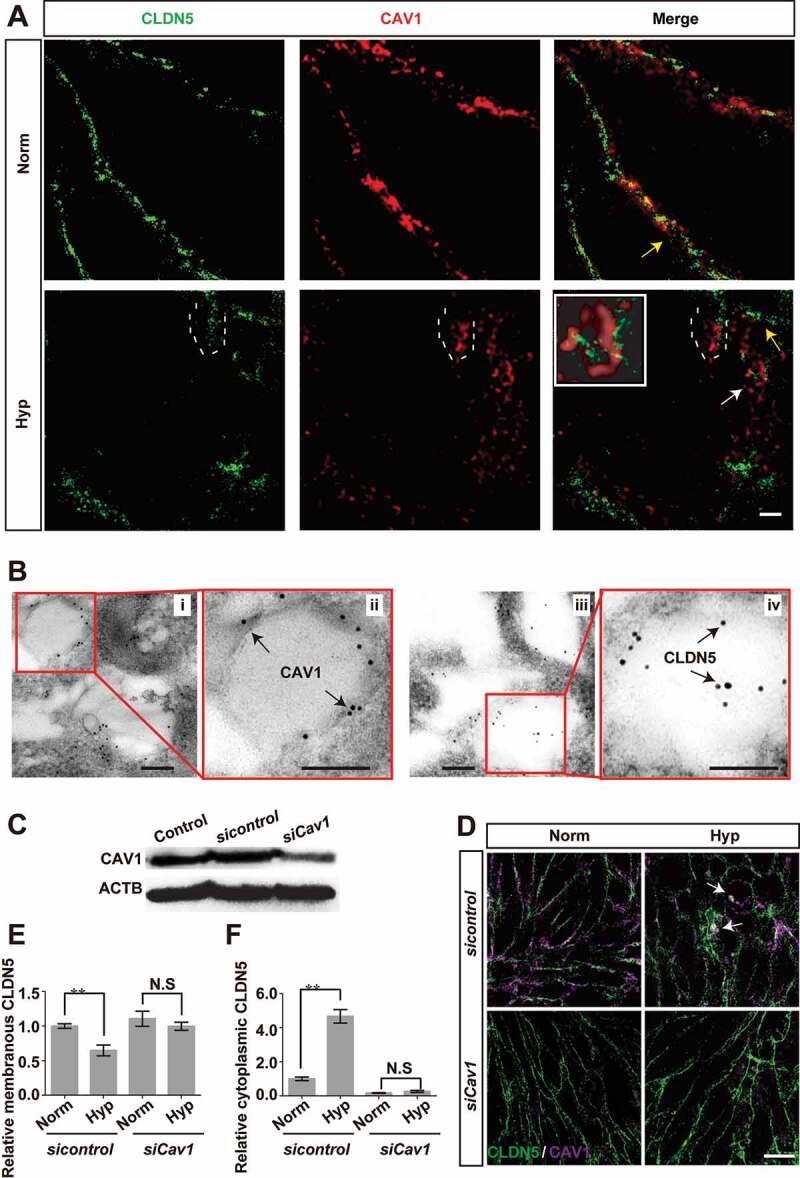
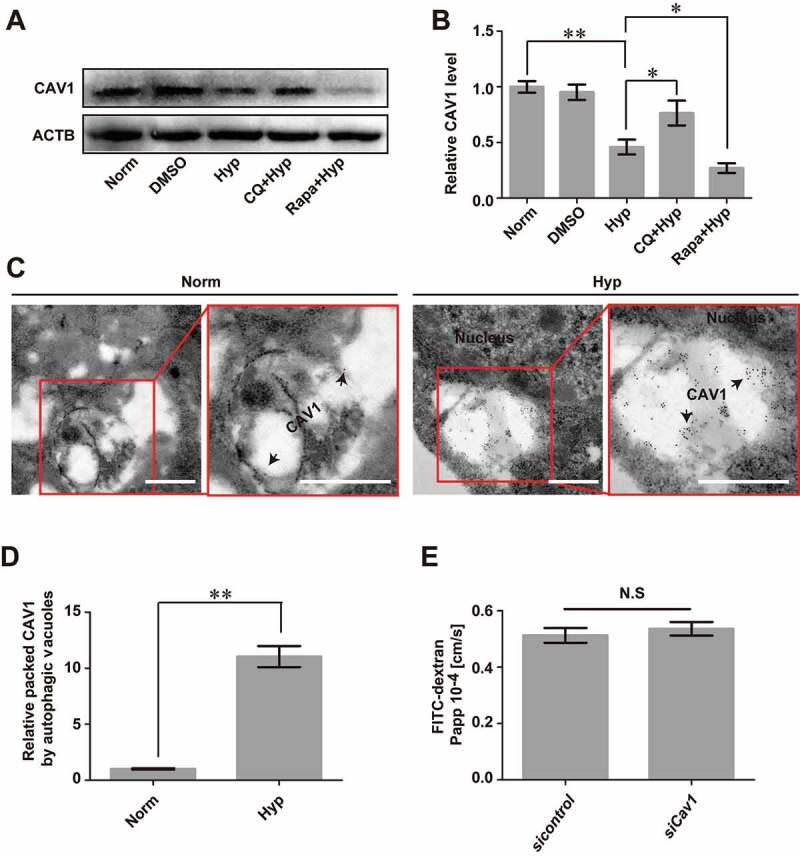
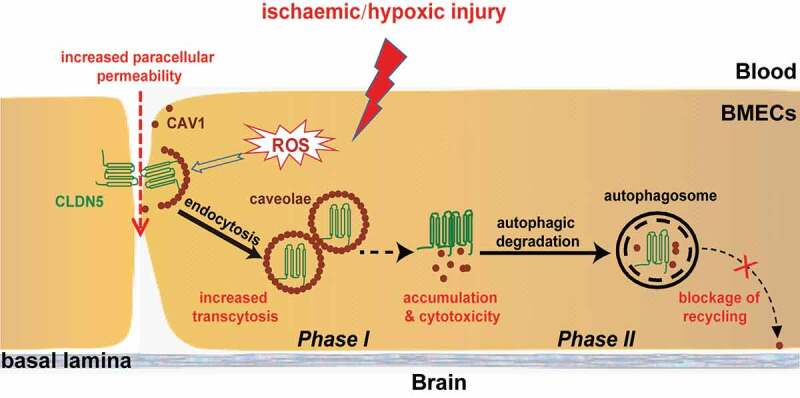
Blood-brain barrier (BBB) disruption is a key event in triggering secondary damage to the central nervous system (CNS) under stroke, and is frequently associated with abnormal macroautophagy/autophagy in brain microvascular endothelial cells (BMECs). However, the underlying mechanism of autophagy in maintaining BBB integrity remains unclear. Here we report that in BMECs of patients suffering stroke, CLDN5 (claudin 5) abnormally aggregates in the cytosol accompanied by autophagy activation. In vivo zebrafish and in vitro cell studies reveal that BBB breakdown is partially caused by CAV1 (caveolin 1)-mediated redistribution of membranous CLDN5 into the cytosol under hypoxia. Meanwhile, autophagy is activated and contributes mainly to the degradation of CAV1 and aggregated CLDN5 in the cytosol of BMECs, therefore alleviating BBB breakdown. Blockage of autophagy by genetic methods or chemicals aggravates cytosolic aggregation of CLDN5, resulting in severer BBB impairment. These data demonstrate that autophagy functions in the protection of BBB integrity by regulating CLDN5 redistribution and provide a potential therapeutic strategy for BBB disorder-related cerebrovascular disease.Abbreviations: BBB: blood-brain barrier; BECN1: beclin 1; BMEC: brain microvascular endothelial cell; CAV1: caveolin 1; CCA: common carotid artery; CLDN5: claudin 5; CNS: central nervous system; CQ: chloroquine; HIF1A: hypoxia inducible factor 1 subunit alpha; MCAO: middle cerebral artery occlusion-reperfusion; OCLN: occludin; ROS: reactive oxygen species; STED: stimulated emission depletion; TEER: trans-endothelial electrical resistance; TEM: transmission electron microscopy; TJ: tight junction; TJP1: tight junction protein 1; UPS: ubiquitin-proteasome system.
Keywords: Autophagy; blood-brain barrier; claudin 5; hypoxia; zebrafish.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
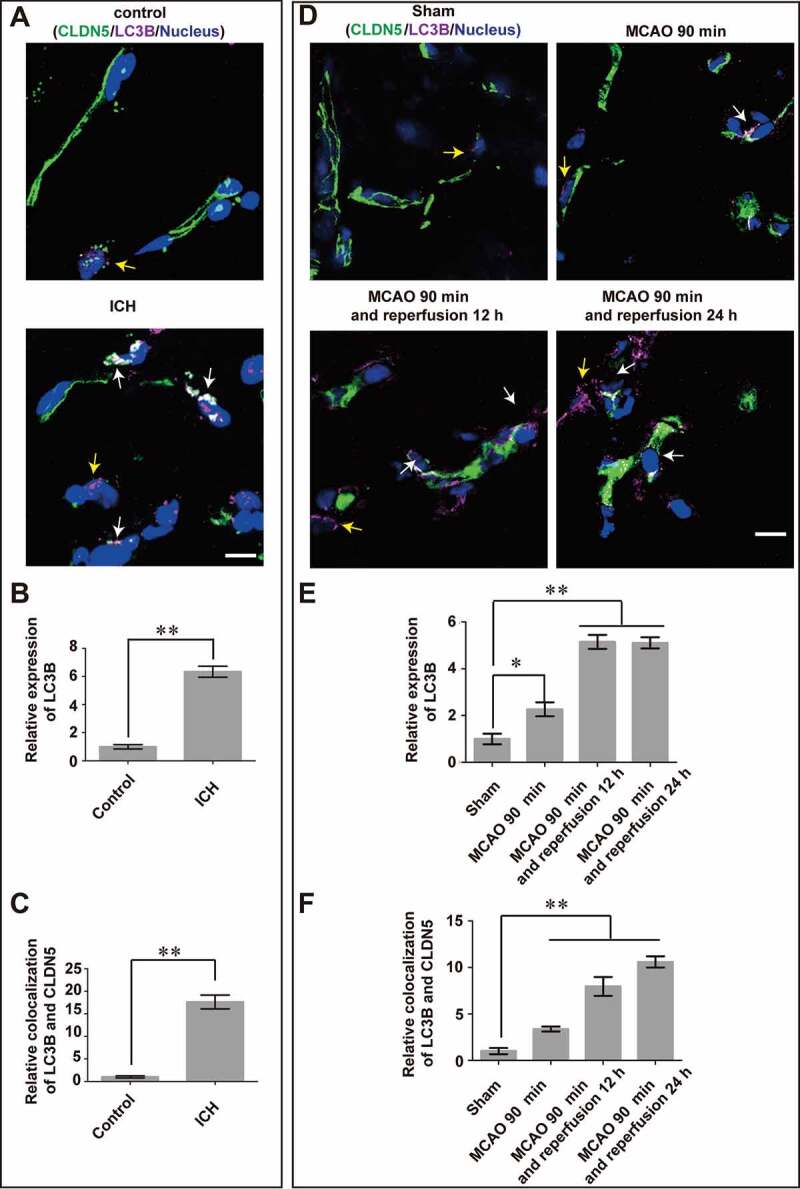
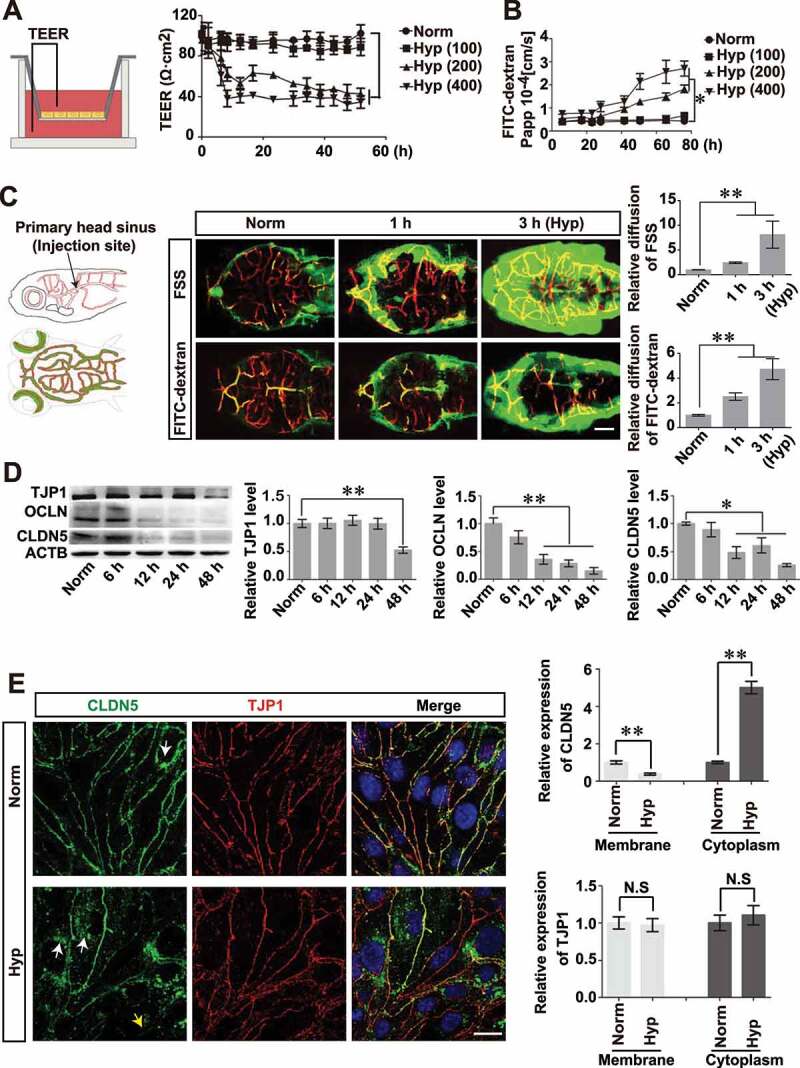
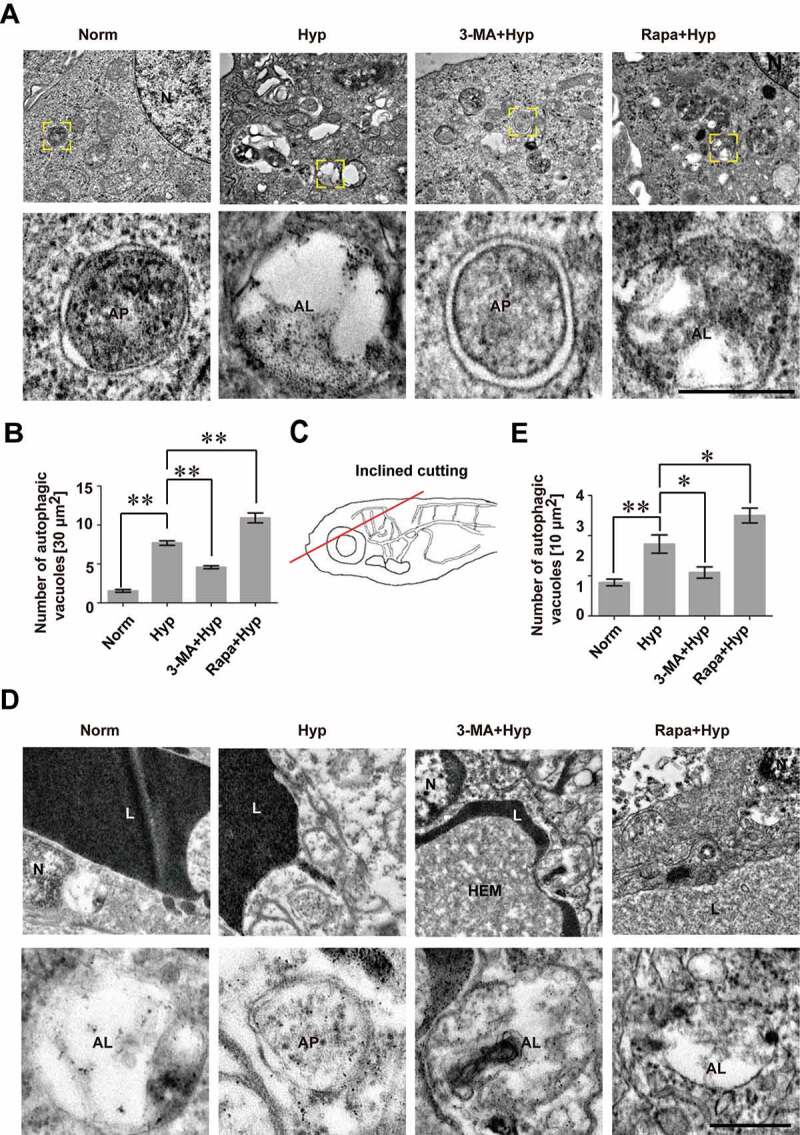
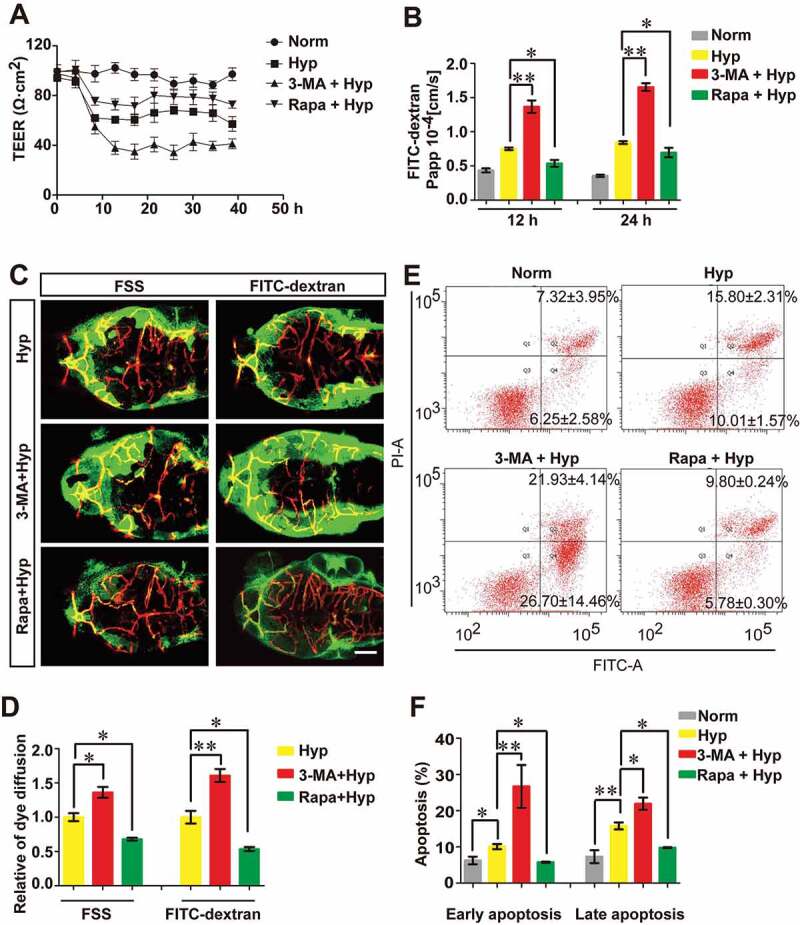
References
-
- Abbott N, Ronnback L, Hansson E.. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7(1):41–53. - PubMed
-
- Liao Z, Yang Z, Piontek A, et al. Specific binding of a mutated fragment of Clostridium perfringens enterotoxin to endothelial claudin-5 and its modulation of cerebral vascular permeability. Neuroscience. 2016;327:53–63. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous