

Elucidation of the miR164c-Guided Gene/Protein Interaction Network Controlling Seed Vigor in Rice
- PMID: 33281848
- PMCID: PMC7688992
- DOI: 10.3389/fpls.2020.589005
Elucidation of the miR164c-Guided Gene/Protein Interaction Network Controlling Seed Vigor in Rice
Abstract
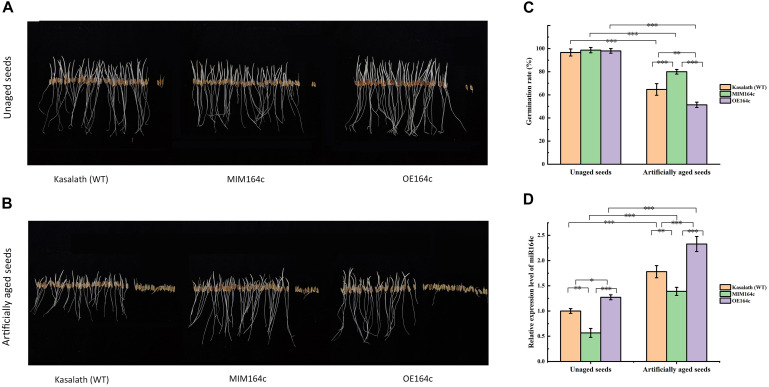
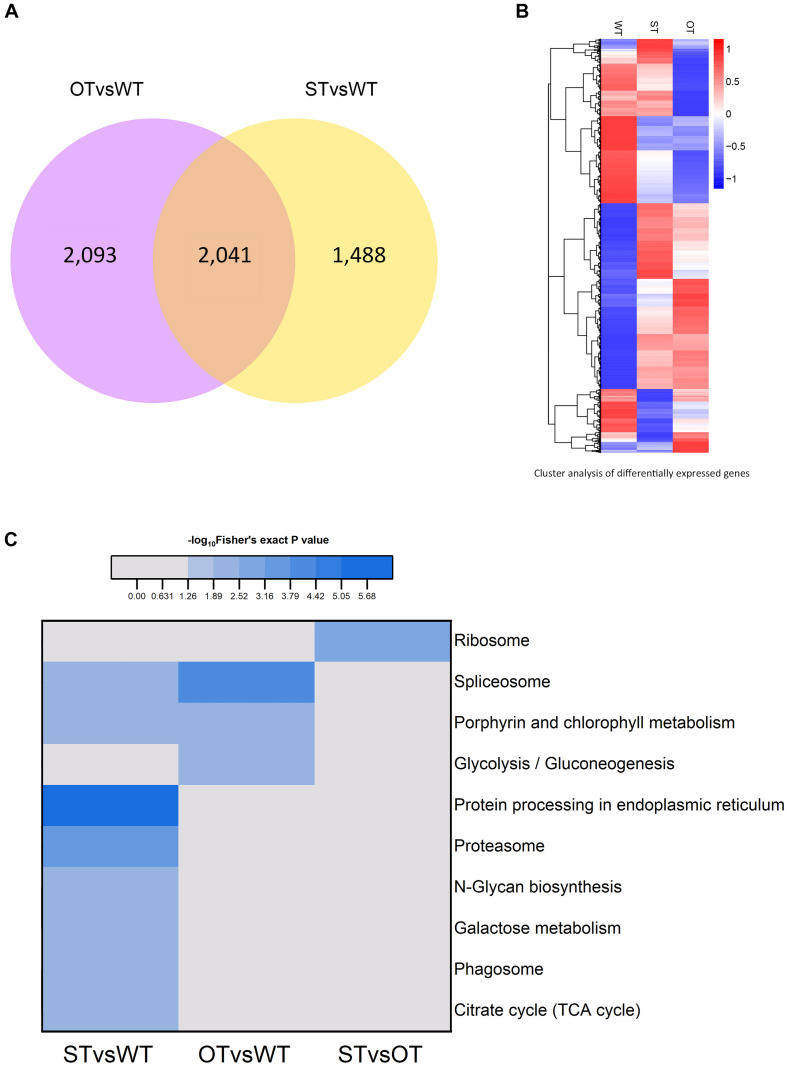
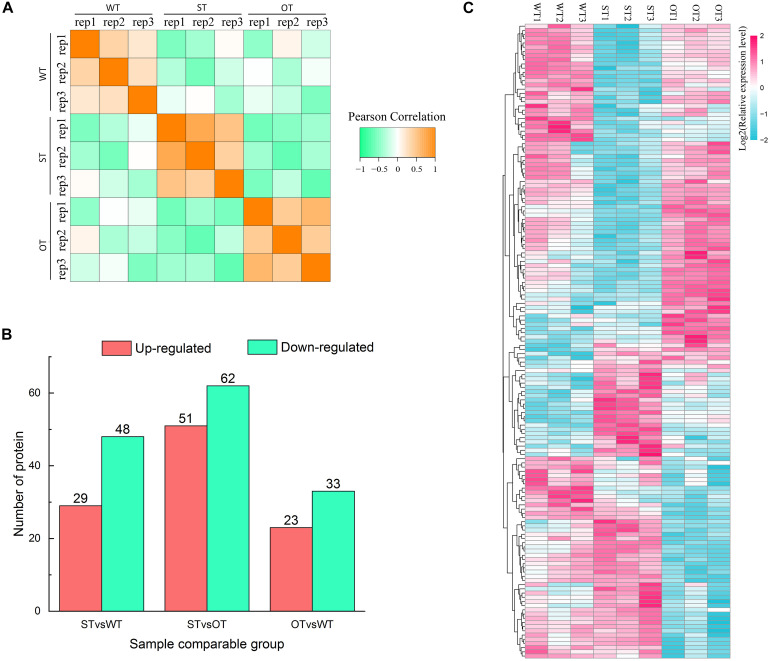
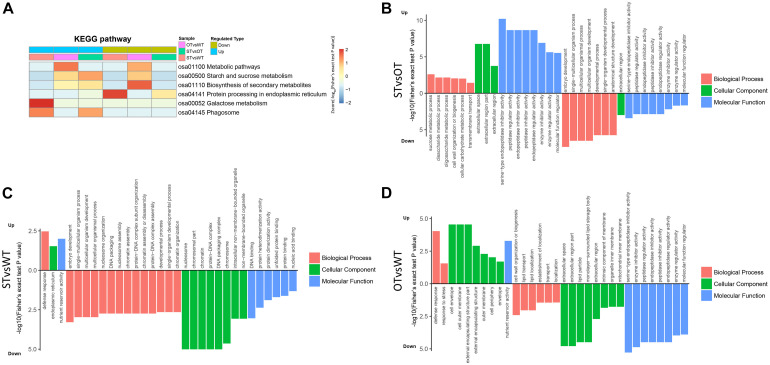
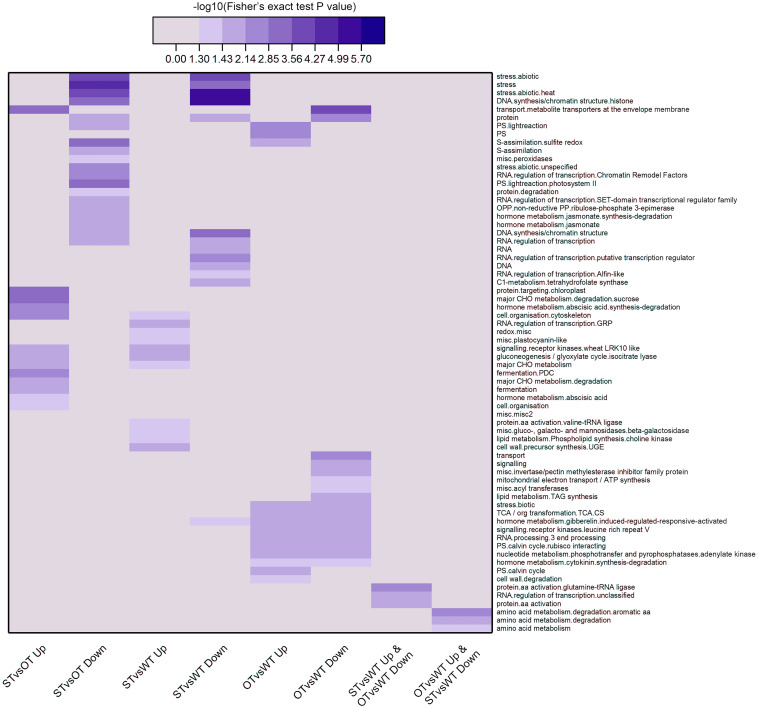
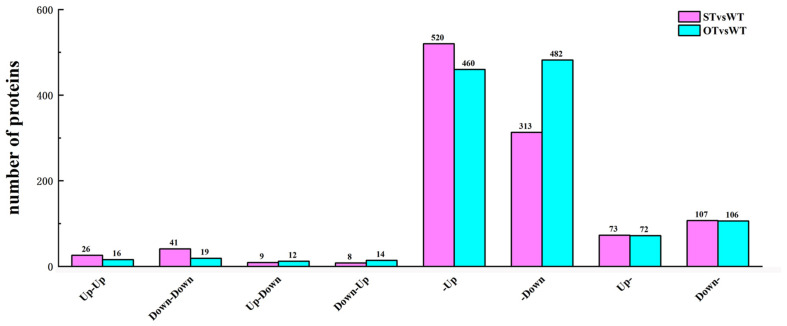
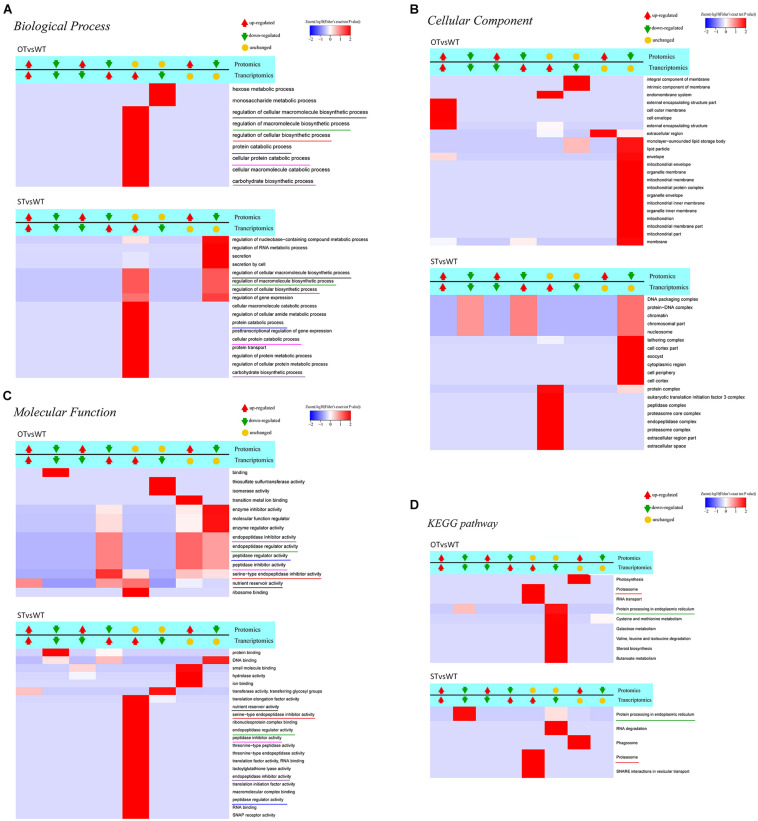
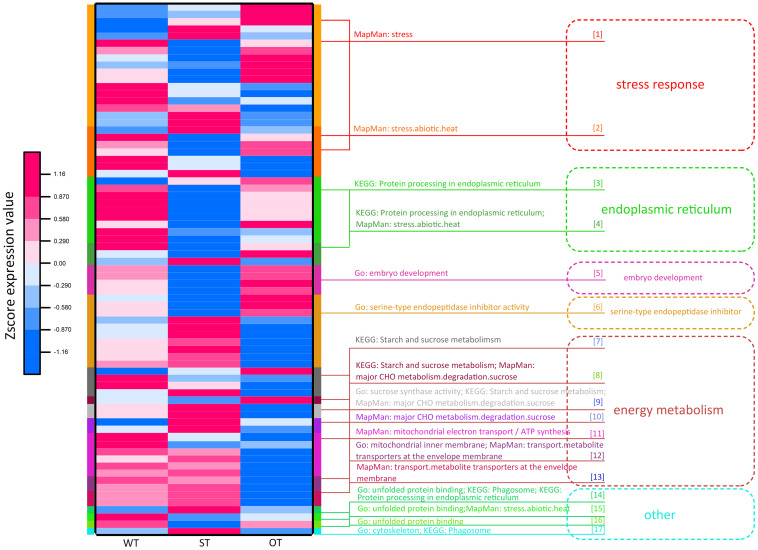
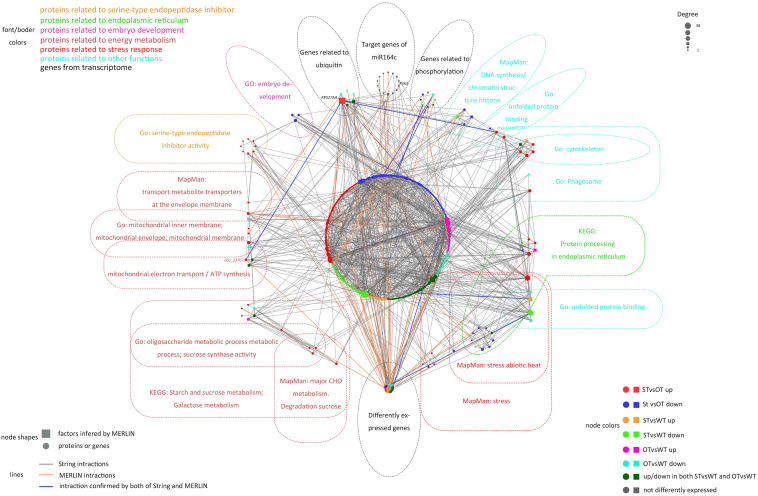
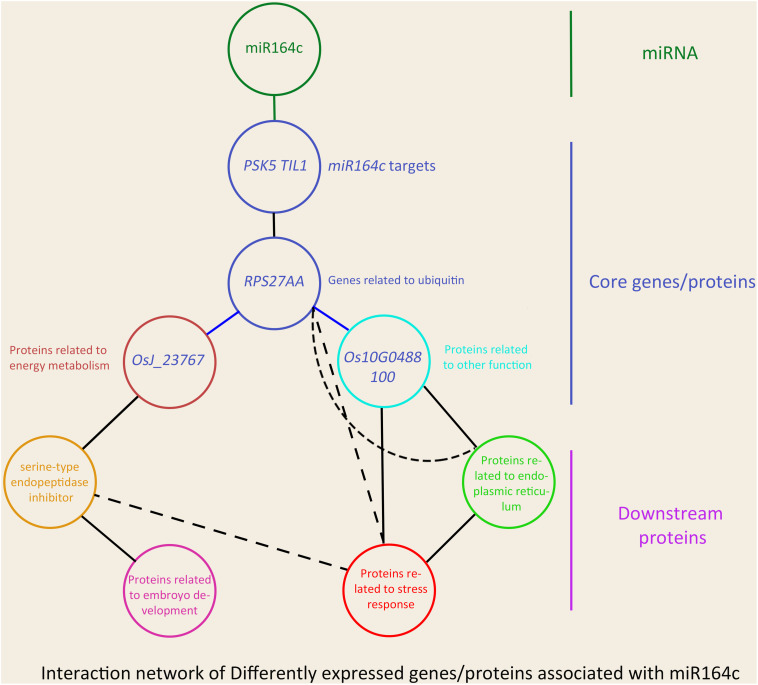
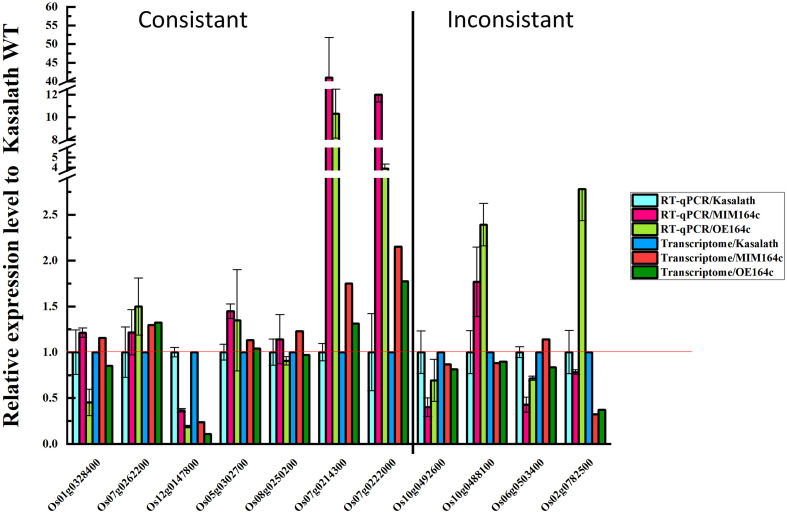
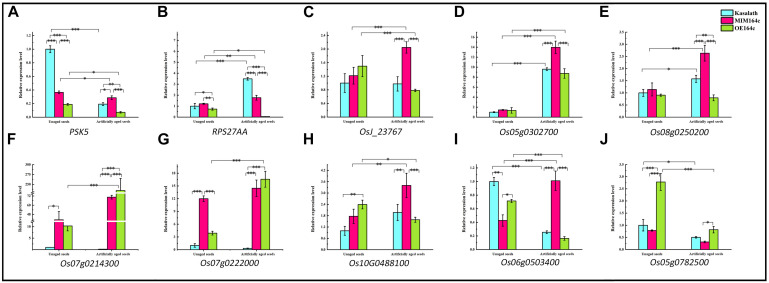
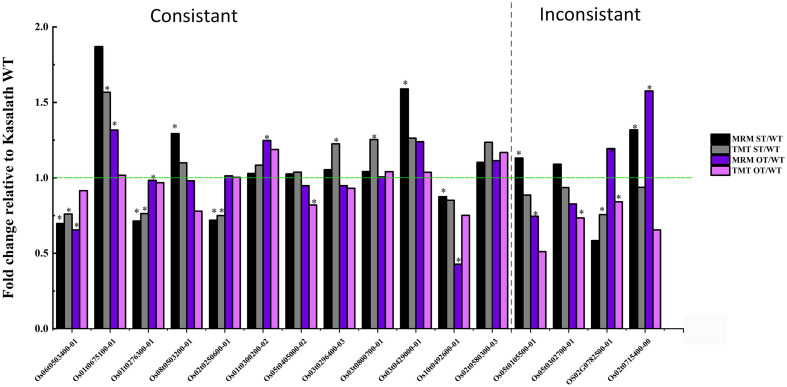
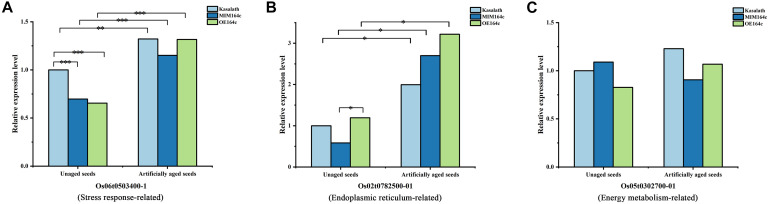
MicroRNAs (miRNAs) play important roles in various aspects of plant physiology and metabolism. The expression level of miR164c is negatively correlated with seed vigor in rice (Oryza sativa L.); however, the mechanism of seed vigor regulation by miR164c remains unknown. Anti-aging capacity is an important indicator of seed vigor. Here, we report an miR164c-guided gene/protein interaction network that regulates the anti-aging ability of rice seeds. Seeds of the wild-type (WT) rice cultivar "Kasalath" and its transgenic derivatives, miR164c-silenced line (MIM164c) and miR164c overexpression line (OE164c), with significant differences in anti-aging capacity, showed significant differences in gene and protein expression levels. The differentially expressed genes (DEGs) or proteins were significantly enriched in six metabolic functional categories related to seed vigor, including "stress response," "protein processing in endoplasmic reticulum (ER)," "embryo development," "serine-type endopeptidase inhibitor," "energy metabolism," and "other." Differences in the expression levels of genes or proteins related to energy metabolism, serine endopeptidase, and stress response in seeds under normal storage conditions may be associated with anti-aging capacity. The results of gene/protein interaction analyses suggest that miR164c first targets PSK5, and the PSK5 protein then interacts with the ubiquitin-associated gene RPS27AA, which simultaneously impacts the genes/proteins in the six above-mentioned functional categories. Expression levels of some of the key genes and proteins in the interaction network were verified by real-time fluorescence quantitative PCR (RT-qPCR) and multiple reaction monitoring mass spectrometry (MRM-MS), respectively. Thus, the present study provides new insights into the miRNA-mediated gene and protein interaction network that regulates seed vigor.
Keywords: Oryza sativa L.; miR164c; proteome; regulatory network; seed vigor; transcriptome.
Copyright © 2020 Huang, Zhou, Shen, Zhou, Wang and Jiang.
Figures
References
-
- Aragão V. P. M., Trindade B. M. C., Reis R. S., Silveira V., Santa-Catarina C. (2019). Storage time affects the germination and proteomic profile of seeds of Cariniana legalis (Mart.) O. Kuntze (Lecythidaceae), an endangered tree species native to the Brazilian Atlantic Forest. Braz. J. Bot. 42 407–419. 10.1007/s40415-019-00551-0 - DOI
-
- Arc E., Ogé L., Grappin P., Rajjou L. (2011). Plant Seed: A Relevant Model to Study Aging Processes. Trivandrum: Transworld Research Network.
LinkOut - more resources
Full Text Sources