

Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease
- PMID: 33283990
- PMCID: PMC7962145
- DOI: 10.1056/NEJMoa2029392
Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease
Abstract
Background: Sickle cell disease is characterized by hemolytic anemia, pain, and progressive organ damage. A high level of erythrocyte fetal hemoglobin (HbF) comprising α- and γ-globins may ameliorate these manifestations by mitigating sickle hemoglobin polymerization and erythrocyte sickling. BCL11A is a repressor of γ-globin expression and HbF production in adult erythrocytes. Its down-regulation is a promising therapeutic strategy for induction of HbF.
Methods: We enrolled patients with sickle cell disease in a single-center, open-label pilot study. The investigational therapy involved infusion of autologous CD34+ cells transduced with the BCH-BB694 lentiviral vector, which encodes a short hairpin RNA (shRNA) targeting BCL11A mRNA embedded in a microRNA (shmiR), allowing erythroid lineage-specific knockdown. Patients were assessed for primary end points of engraftment and safety and for hematologic and clinical responses to treatment.
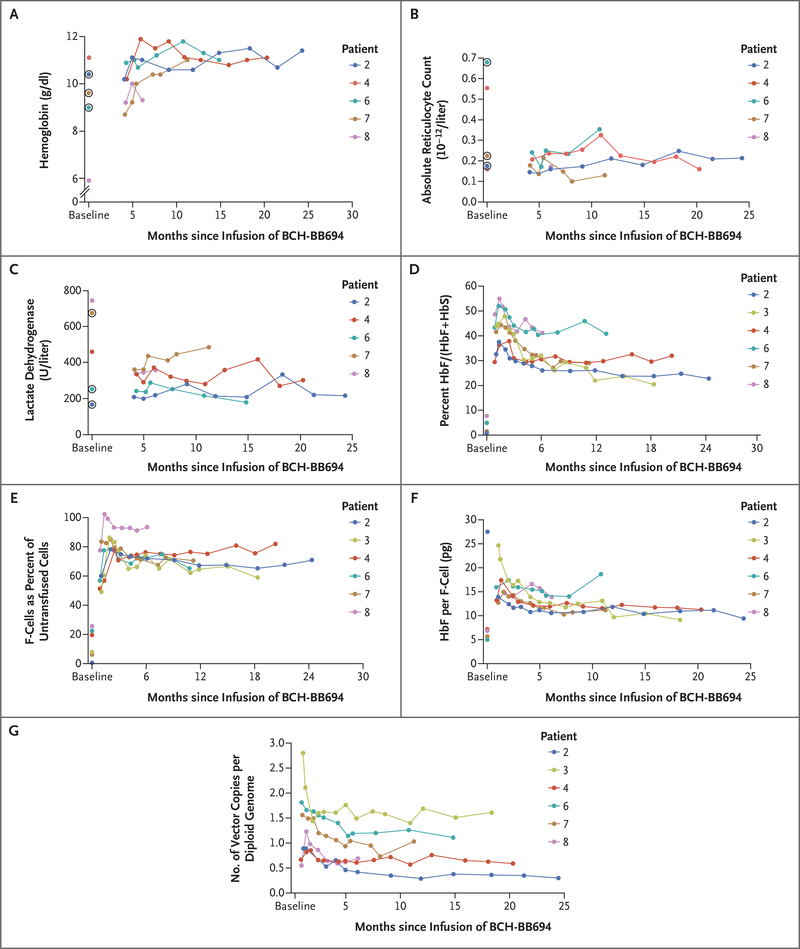
Results: As of October 2020, six patients had been followed for at least 6 months after receiving BCH-BB694 gene therapy; median follow-up was 18 months (range, 7 to 29). All patients had engraftment, and adverse events were consistent with effects of the preparative chemotherapy. All the patients who could be fully evaluated achieved robust and stable HbF induction (percentage HbF/(F+S) at most recent follow-up, 20.4 to 41.3%), with HbF broadly distributed in red cells (F-cells 58.9 to 93.6% of untransfused red cells) and HbF per F-cell of 9.0 to 18.6 pg per cell. Clinical manifestations of sickle cell disease were reduced or absent during the follow-up period.
Conclusions: This study validates BCL11A inhibition as an effective target for HbF induction and provides preliminary evidence that shmiR-based gene knockdown offers a favorable risk-benefit profile in sickle cell disease. (Funded by the National Institutes of Health; ClinicalTrials.gov number, NCT03282656).
Copyright © 2020 Massachusetts Medical Society.
Figures
Comment in
-
CRISPR gene therapy shows promise against blood diseases.Nature. 2020 Dec;588(7838):383. doi: 10.1038/d41586-020-03476-x. Nature. 2020. PMID: 33299166 No abstract available.
-
Induction of Fetal Hemoglobin by Gene Therapy.N Engl J Med. 2021 Jan 21;384(3):284-285. doi: 10.1056/NEJMe2034338. N Engl J Med. 2021. PMID: 33471981 No abstract available.
References
-
- Krishnamurti L, Abel S, Maiers M, Flesch S. Availability of unrelated donors for hematopoietic stem cell transplantation for hemoglobinopathies. Bone Marrow Transplant 2003;31:547–50. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical