

Improving the informativeness of Mendelian disease-derived pathogenicity scores for common disease
- PMID: 33288751
- PMCID: PMC7721881
- DOI: 10.1038/s41467-020-20087-2
Improving the informativeness of Mendelian disease-derived pathogenicity scores for common disease
Abstract
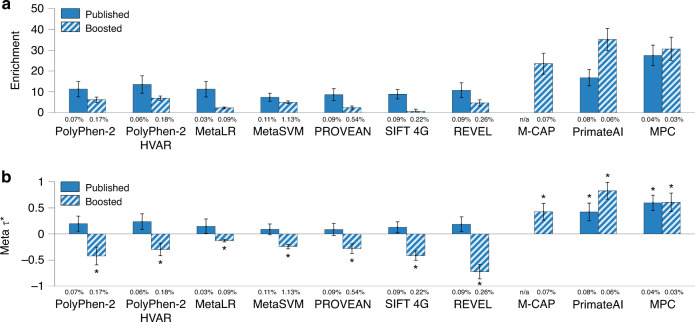
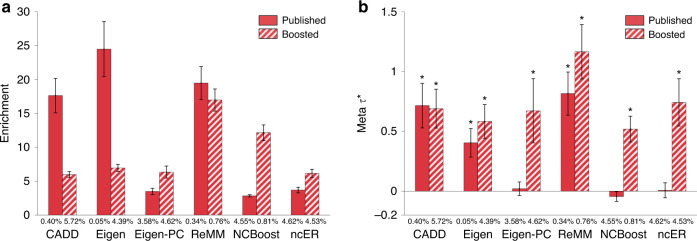
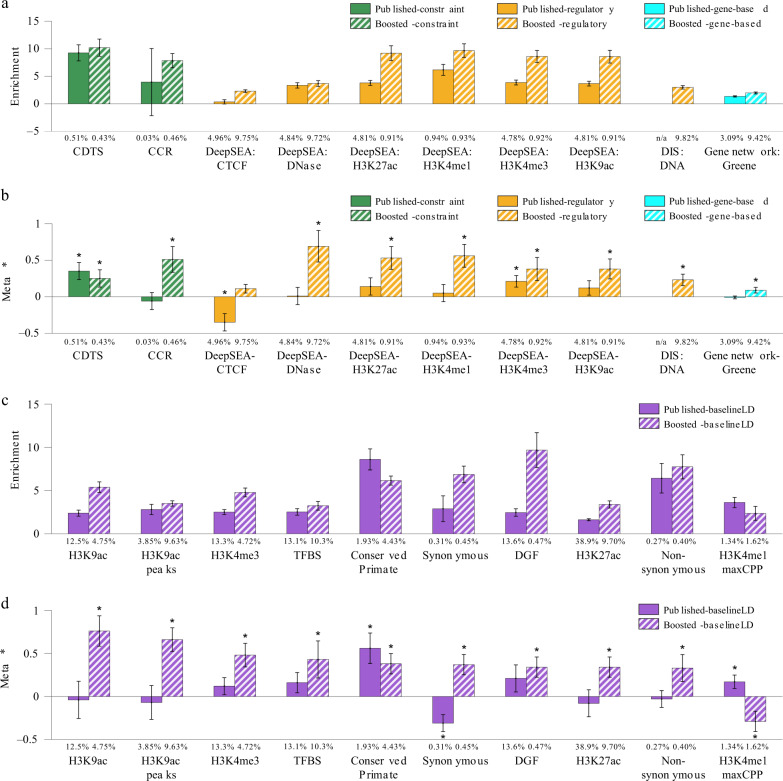
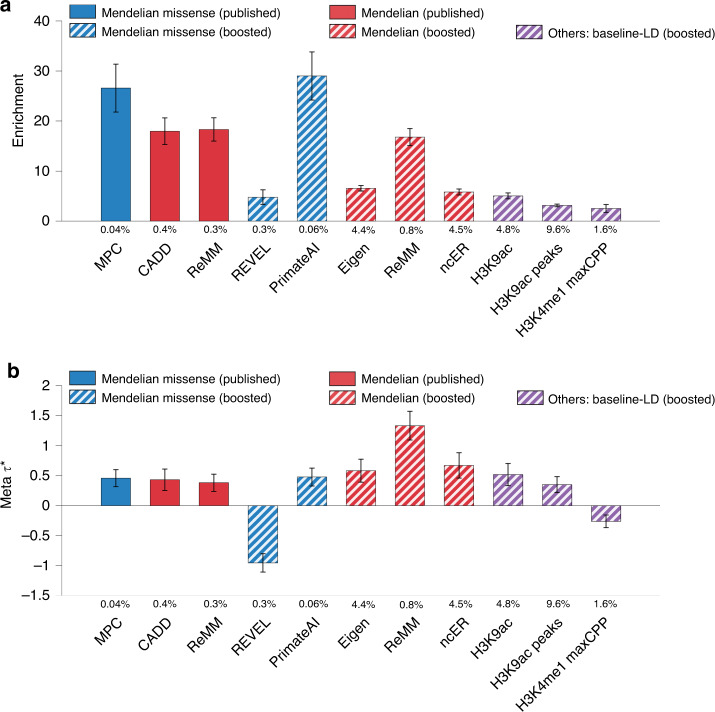
Despite considerable progress on pathogenicity scores prioritizing variants for Mendelian disease, little is known about the utility of these scores for common disease. Here, we assess the informativeness of Mendelian disease-derived pathogenicity scores for common disease and improve upon existing scores. We first apply stratified linkage disequilibrium (LD) score regression to evaluate published pathogenicity scores across 41 common diseases and complex traits (average N = 320K). Several of the resulting annotations are informative for common disease, even after conditioning on a broad set of functional annotations. We then improve upon published pathogenicity scores by developing AnnotBoost, a machine learning framework to impute and denoise pathogenicity scores using a broad set of functional annotations. AnnotBoost substantially increases the informativeness for common disease of both previously uninformative and previously informative pathogenicity scores, implying that Mendelian and common disease variants share similar properties. The boosted scores also produce improvements in heritability model fit and in classifying disease-associated, fine-mapped SNPs. Our boosted scores may improve fine-mapping and candidate gene discovery for common disease.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
