

Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads
- PMID: 33288906
- PMCID: PMC7954704
- DOI: 10.1038/s41587-020-0719-5
Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads
Abstract
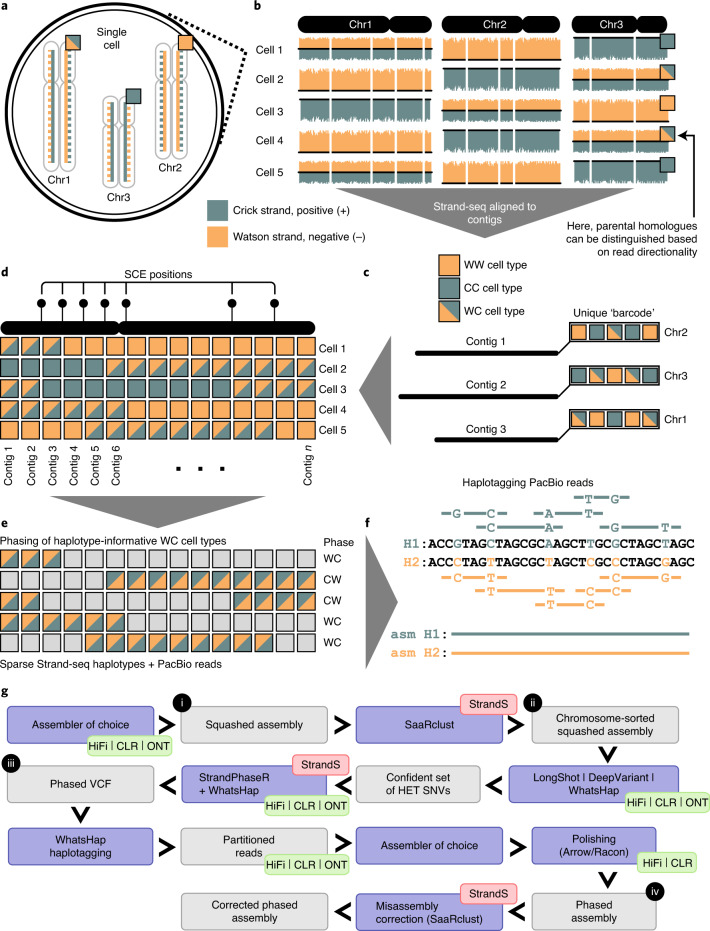
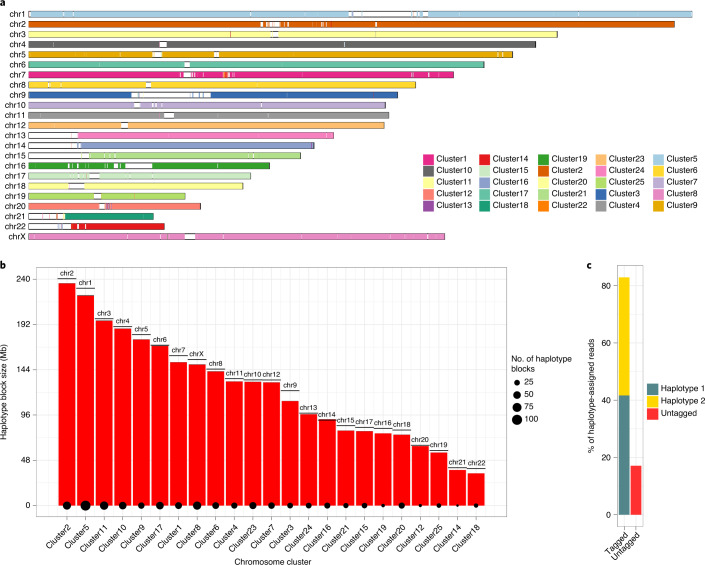
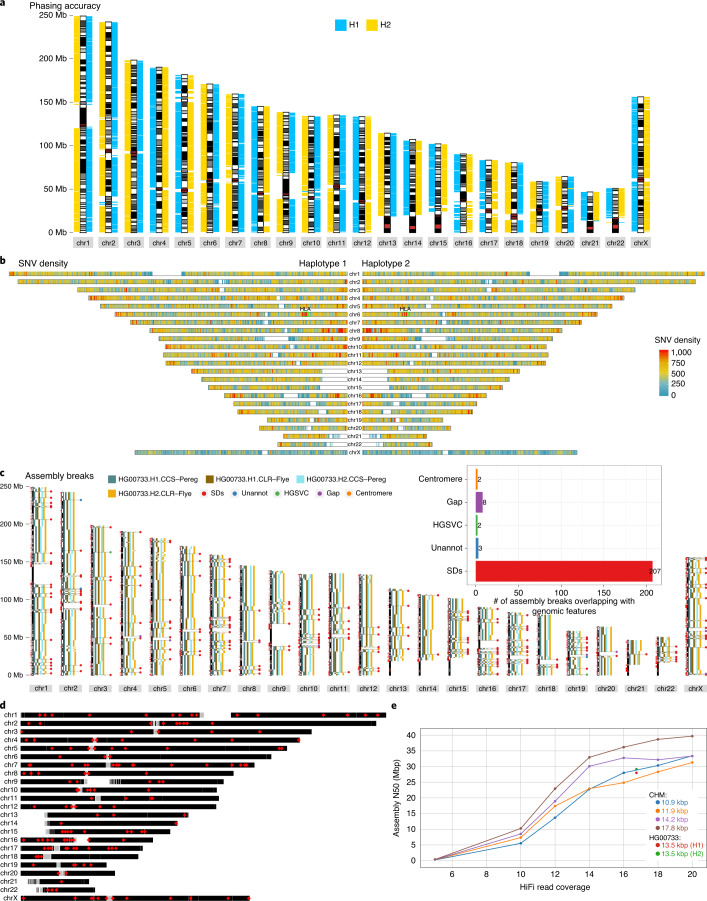
Human genomes are typically assembled as consensus sequences that lack information on parental haplotypes. Here we describe a reference-free workflow for diploid de novo genome assembly that combines the chromosome-wide phasing and scaffolding capabilities of single-cell strand sequencing1,2 with continuous long-read or high-fidelity3 sequencing data. Employing this strategy, we produced a completely phased de novo genome assembly for each haplotype of an individual of Puerto Rican descent (HG00733) in the absence of parental data. The assemblies are accurate (quality value > 40) and highly contiguous (contig N50 > 23 Mbp) with low switch error rates (0.17%), providing fully phased single-nucleotide variants, indels and structural variants. A comparison of Oxford Nanopore Technologies and Pacific Biosciences phased assemblies identified 154 regions that are preferential sites of contig breaks, irrespective of sequencing technology or phasing algorithms.
Conflict of interest statement
E.E.E. was on the scientific advisory board of DNAnexus (2012–2020).
Figures
References
Publication types
MeSH terms
Grants and funding
- R01 HG002898/HG/NHGRI NIH HHS/United States
- U01 HG010961/HG/NHGRI NIH HHS/United States
- U41 HG010972/HG/NHGRI NIH HHS/United States
- T32 LM012419/LM/NLM NIH HHS/United States
- U41 HG007497/HG/NHGRI NIH HHS/United States
- T32 HG008345/HG/NHGRI NIH HHS/United States
- U01 HG010971/HG/NHGRI NIH HHS/United States
- R01 HG010485/HG/NHGRI NIH HHS/United States
- U24 HG007497/HG/NHGRI NIH HHS/United States
- R01 HG002385/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- UM1 HG010971/HG/NHGRI NIH HHS/United States
- P30 CA034196/CA/NCI NIH HHS/United States
- T32 HG000035/HG/NHGRI NIH HHS/United States
- R01 HG010169/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
