

MicroRNA miR-24-3p reduces DNA damage responses, apoptosis, and susceptibility to chronic obstructive pulmonary disease
- PMID: 33290275
- PMCID: PMC7934877
- DOI: 10.1172/jci.insight.134218
MicroRNA miR-24-3p reduces DNA damage responses, apoptosis, and susceptibility to chronic obstructive pulmonary disease
Abstract
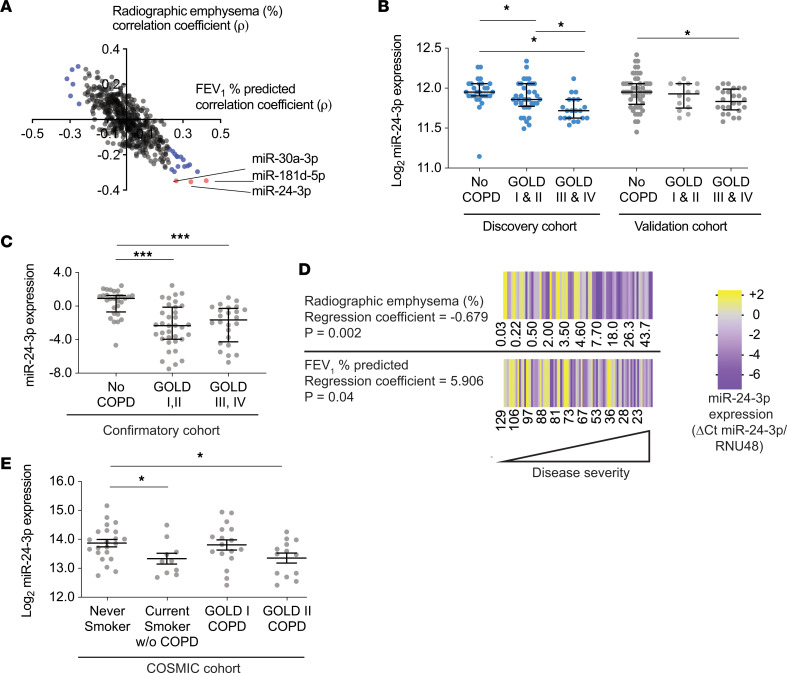
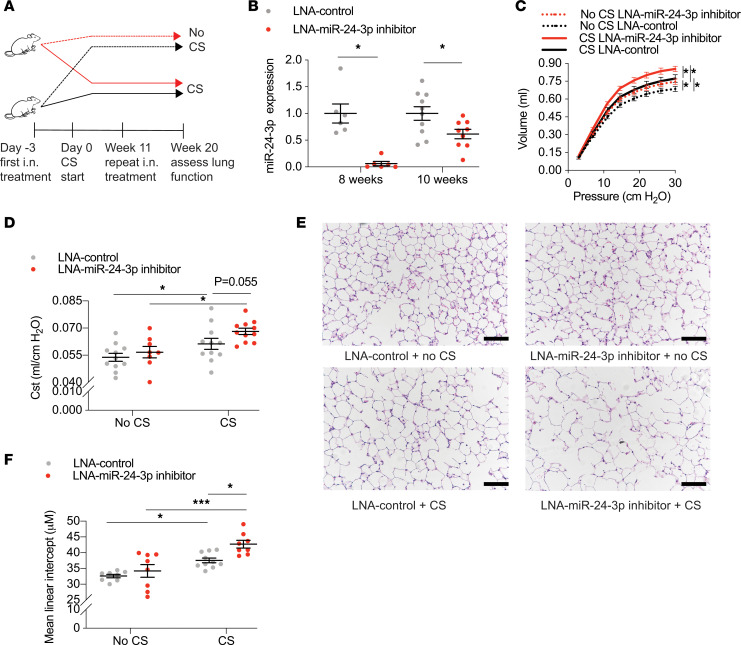
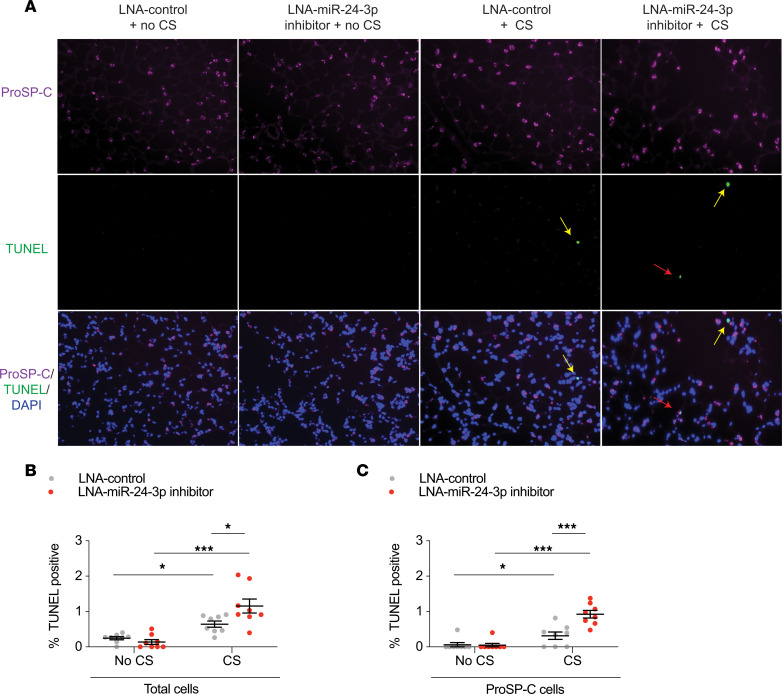
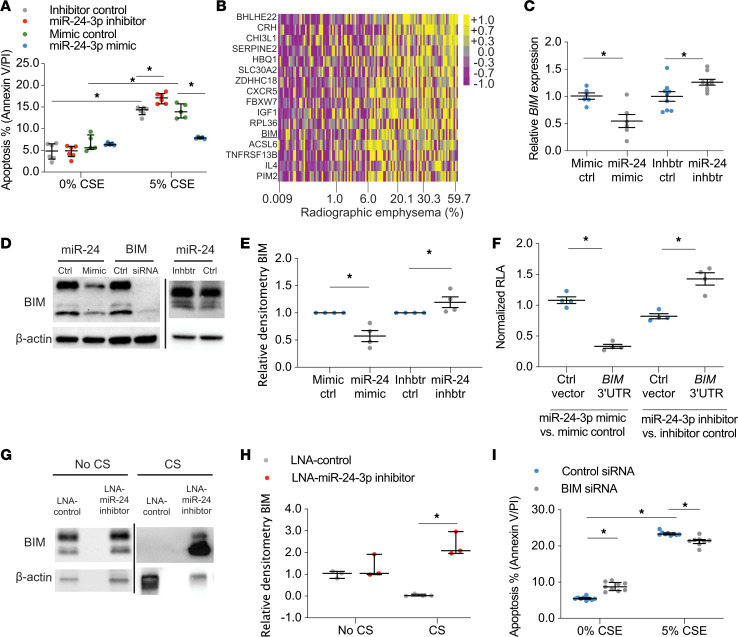
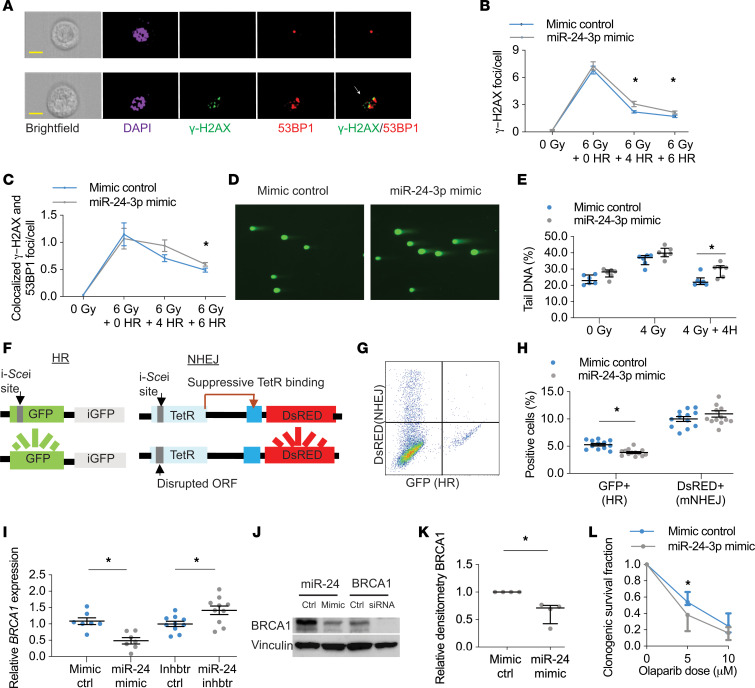
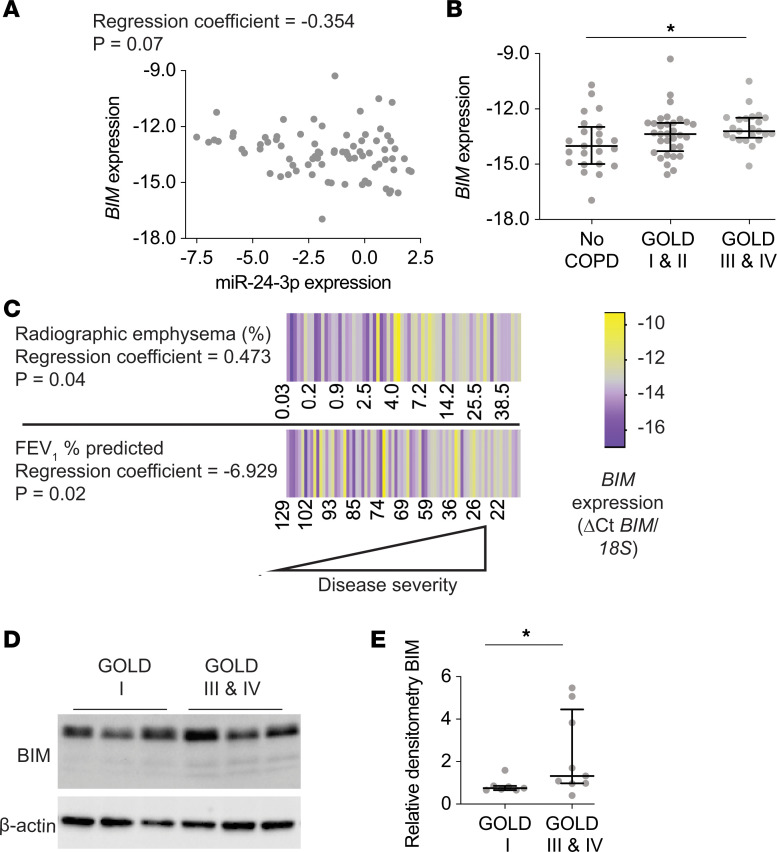
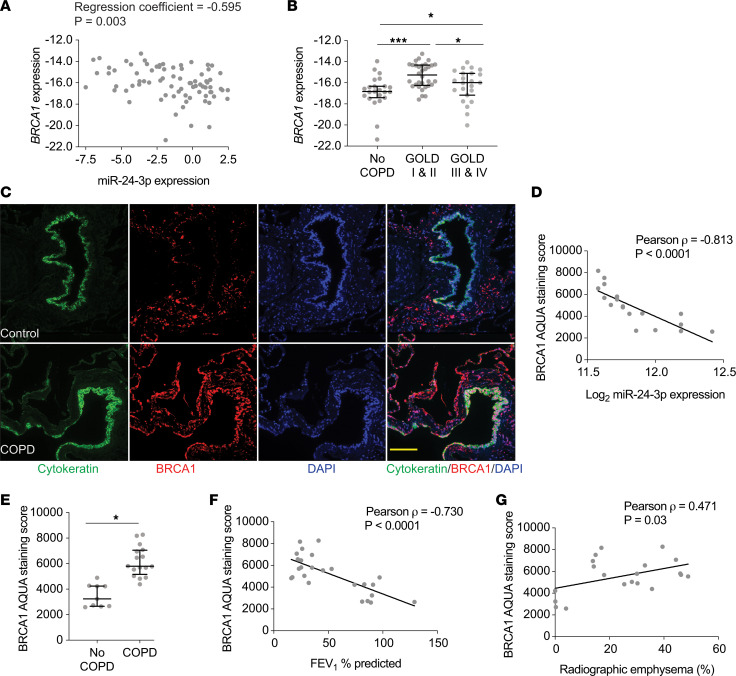
The pathogenesis of chronic obstructive pulmonary disease (COPD) involves aberrant responses to cellular stress caused by chronic cigarette smoke (CS) exposure. However, not all smokers develop COPD and the critical mechanisms that regulate cellular stress responses to increase COPD susceptibility are not understood. Because microRNAs are well-known regulators of cellular stress responses, we evaluated microRNA expression arrays performed on distal parenchymal lung tissue samples from 172 subjects with and without COPD. We identified miR-24-3p as the microRNA that best correlated with radiographic emphysema and validated this finding in multiple cohorts. In a CS exposure mouse model, inhibition of miR-24-3p increased susceptibility to apoptosis, including alveolar type II epithelial cell apoptosis, and emphysema severity. In lung epithelial cells, miR-24-3p suppressed apoptosis through the BH3-only protein BIM and suppressed homology-directed DNA repair and the DNA repair protein BRCA1. Finally, we found BIM and BRCA1 were increased in COPD lung tissue, and BIM and BRCA1 expression inversely correlated with miR-24-3p. We concluded that miR-24-3p, a regulator of the cellular response to DNA damage, is decreased in COPD, and decreased miR-24-3p increases susceptibility to emphysema through increased BIM and apoptosis.
Keywords: Apoptosis; COPD; DNA repair; Pulmonology.
Conflict of interest statement
Figures
Similar articles
-
MiR-223 is increased in lungs of patients with COPD and modulates cigarette smoke-induced pulmonary inflammation.Am J Physiol Lung Cell Mol Physiol. 2021 Dec 1;321(6):L1091-L1104. doi: 10.1152/ajplung.00252.2021. Epub 2021 Oct 20. Am J Physiol Lung Cell Mol Physiol. 2021. PMID: 34668437
-
MicroRNA-221-3p alleviates cell apoptosis and inflammatory response by targeting cyclin dependent kinase inhibitor 1B in chronic obstructive pulmonary disease.Bioengineered. 2021 Dec;12(1):5705-5715. doi: 10.1080/21655979.2021.1967837. Bioengineered. 2021. PMID: 34516316 Free PMC article.
-
MicroRNA-150 protects against cigarette smoke-induced lung inflammation and airway epithelial cell apoptosis through repressing p53: MicroRNA-150 in CS-induced lung inflammation.Hum Exp Toxicol. 2018 Sep;37(9):920-928. doi: 10.1177/0960327117741749. Epub 2017 Dec 5. Hum Exp Toxicol. 2018. PMID: 29205062
-
DNA repair as an emerging target for COPD-lung cancer overlap.Respir Investig. 2019 Mar;57(2):111-121. doi: 10.1016/j.resinv.2018.11.005. Epub 2019 Jan 7. Respir Investig. 2019. PMID: 30630751 Review.
-
Bringing Light to Chronic Obstructive Pulmonary Disease Pathogenesis and Resilience.Ann Am Thorac Soc. 2018 Dec;15(Suppl 4):S227-S233. doi: 10.1513/AnnalsATS.201808-583MG. Ann Am Thorac Soc. 2018. PMID: 30759011 Free PMC article. Review.
Cited by
-
Implications of DNA damage in chronic lung disease.Front Cell Dev Biol. 2024 Oct 31;12:1436767. doi: 10.3389/fcell.2024.1436767. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 39544366 Free PMC article. Review.
-
The role of miRNAs in alveolar epithelial cells in emphysema.Biomed Pharmacother. 2021 Nov;143:112216. doi: 10.1016/j.biopha.2021.112216. Epub 2021 Sep 27. Biomed Pharmacother. 2021. PMID: 34649347 Free PMC article. Review.
-
Identification and Construction of a R-loop Mediated Diagnostic Model and Associated Immune Microenvironment of COPD through Machine Learning and Single-Cell Transcriptomics.Inflammation. 2025 Aug;48(4):2802-2823. doi: 10.1007/s10753-024-02232-x. Epub 2025 Jan 11. Inflammation. 2025. PMID: 39798034 Free PMC article.
-
MicroRNA-377-3p exacerbates chronic obstructive pulmonary disease through suppressing ZFP36L1 expression and inducing lung fibroblast senescence.Respir Res. 2024 Feb 5;25(1):67. doi: 10.1186/s12931-024-02696-3. Respir Res. 2024. PMID: 38317146 Free PMC article.
-
Characterization of pulmonary vascular remodeling and MicroRNA-126-targets in COPD-pulmonary hypertension.Respir Res. 2022 Dec 15;23(1):349. doi: 10.1186/s12931-022-02267-4. Respir Res. 2022. PMID: 36522710 Free PMC article.
References
-
- GBD 2015 Chronic Respiratory Disease Collaborators Global, regional, and national deaths, prevalence, disability-adjusted life years, and years lived with disability for chronic obstructive pulmonary disease and asthma, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Respir Med. 2017;5(9):691–706. doi: 10.1016/S2213-2600(17)30293-X. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- K08 HL135402/HL/NHLBI NIH HHS/United States
- R21 LM012884/LM/NLM NIH HHS/United States
- R01 HL127349/HL/NHLBI NIH HHS/United States
- UL1 TR001863/TR/NCATS NIH HHS/United States
- P30 AG021342/AG/NIA NIH HHS/United States
- U01 HL122626/HL/NHLBI NIH HHS/United States
- R01 HL138396/HL/NHLBI NIH HHS/United States
- U01 HL145567/HL/NHLBI NIH HHS/United States
- R03 HL154275/HL/NHLBI NIH HHS/United States
- K01 HL125474/HL/NHLBI NIH HHS/United States
- R01 HL153604/HL/NHLBI NIH HHS/United States
- U54 HG008540/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
